

Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy
- PMID: 27164704
- PMCID: PMC4898312
- DOI: 10.1212/WNL.0000000000002740
Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy
Abstract
Objective: To determine the phenotypic spectrum caused by mutations in GRIN1 encoding the NMDA receptor subunit GluN1 and to investigate their underlying functional pathophysiology.
Methods: We collected molecular and clinical data from several diagnostic and research cohorts. Functional consequences of GRIN1 mutations were investigated in Xenopus laevis oocytes.
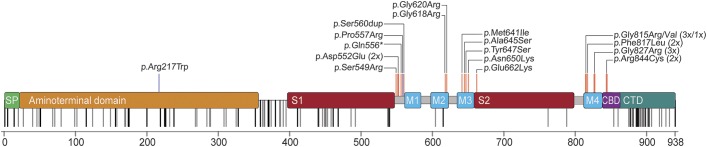
Results: We identified heterozygous de novo GRIN1 mutations in 14 individuals and reviewed the phenotypes of all 9 previously reported patients. These 23 individuals presented with a distinct phenotype of profound developmental delay, severe intellectual disability with absent speech, muscular hypotonia, hyperkinetic movement disorder, oculogyric crises, cortical blindness, generalized cerebral atrophy, and epilepsy. Mutations cluster within transmembrane segments and result in loss of channel function of varying severity with a dominant-negative effect. In addition, we describe 2 homozygous GRIN1 mutations (1 missense, 1 truncation), each segregating with severe neurodevelopmental phenotypes in consanguineous families.
Conclusions: De novo GRIN1 mutations are associated with severe intellectual disability with cortical visual impairment as well as oculomotor and movement disorders being discriminating phenotypic features. Loss of NMDA receptor function appears to be the underlying disease mechanism. The identification of both heterozygous and homozygous mutations blurs the borders of dominant and recessive inheritance of GRIN1-associated disorders.
© 2016 American Academy of Neurology.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases