

Next-generation proteasome inhibitor oprozomib synergizes with modulators of the unfolded protein response to suppress hepatocellular carcinoma
- PMID: 27167000
- PMCID: PMC5085204
- DOI: 10.18632/oncotarget.9222
Next-generation proteasome inhibitor oprozomib synergizes with modulators of the unfolded protein response to suppress hepatocellular carcinoma
Abstract
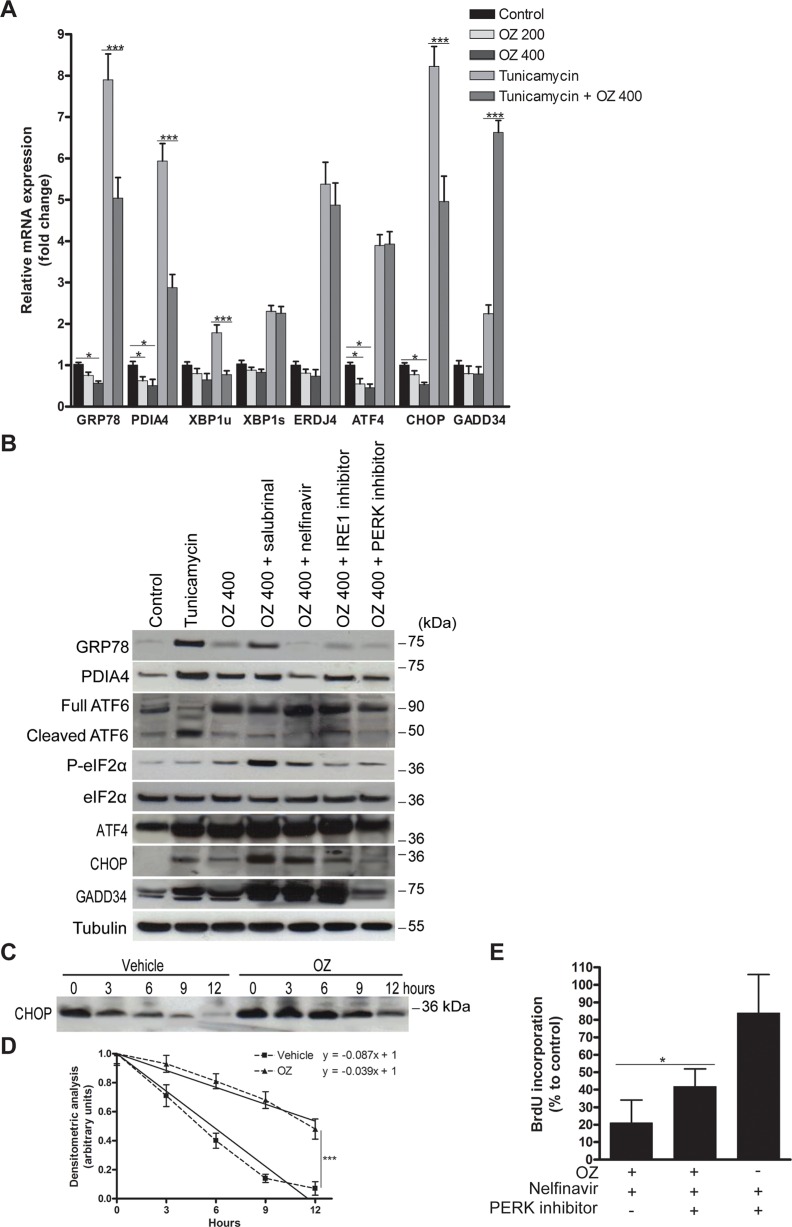
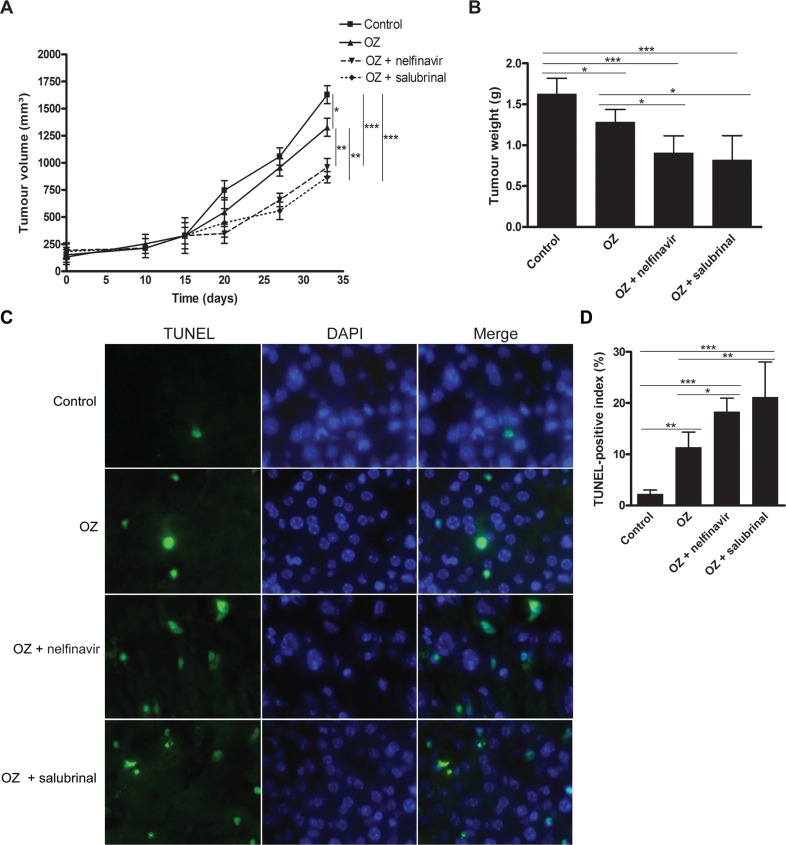
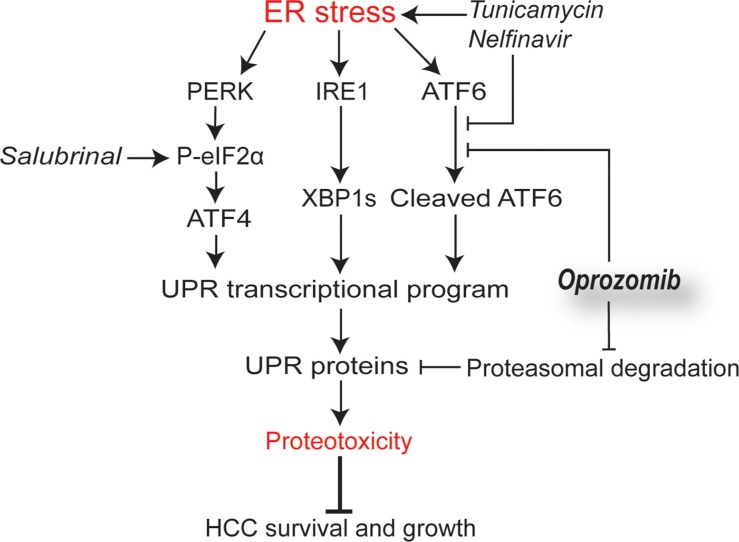
Hepatocellular carcinoma (HCC) responds poorly to conventional systemic therapies. The first-in-class proteasome inhibitor bortezomib has been approved in clinical use for hematologic malignancies and has shown modest activity in solid tumors, including HCC. However, a considerable proportion of patients fail to respond and experience important adverse events. Recently, the next-generation orally bioavailable irreversible proteasome inhibitor oprozomib was developed. Here, we assessed the efficacy of oprozomib and its effects on the unfolded protein response (UPR), a signaling cascade activated through the ATF6, PERK and IRE1 pathways by accumulation of unfolded proteins in the endoplasmic reticulum, in HCC. The effects of oprozomib and the role of the UPR were evaluated in HCC cell lines and in diethylnitrosamine-induced and xenograft mouse models for HCC. Oprozomib dose-dependently reduced the viability and proliferation of human HCC cells. Unexpectedly, oprozomib-treated cells displayed diminished cytoprotective ATF6-mediated signal transduction as well as unaltered PERK and IRE1 signaling. However, oprozomib increased pro-apoptotic UPR-mediated protein levels by prolonging their half-life, implying that the proteasome acts as a negative UPR regulator. Supplementary boosting of UPR activity synergistically improved the sensitivity to oprozomib via the PERK pathway. Oral oprozomib displayed significant antitumor effects in the orthotopic and xenograft models for HCC, and importantly, combining oprozomib with different UPR activators enhanced the antitumor efficacy by stimulating UPR-induced apoptosis without cumulative toxicity. In conclusion, next-generation proteasome inhibition by oprozomib results in dysregulated UPR activation in HCC. This finding can be exploited to enhance the antitumor efficacy by combining oprozomib with clinically applicable UPR activators.
Keywords: endoplasmic reticulum; hepatocellular carcinoma; proteasome inhibitor; stress; unfolded protein response.
Conflict of interest statement
None to declare.
Figures


References
-
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Lyon, France: International Agency for Research on Cancer; 2013. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase [Internet]http://globocan.iarc.fr Available from: accessed on 20/11/2015.
-
- EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma J. Hepatol. 2012;56:908–943. - PubMed
-
- Mateos MV, Ocio EM, San Miguel JF. Novel generation of agents with proven clinical activity in multiple myeloma. Semin. Oncol. 2013;40:618–633. - PubMed
-
- Saeki I, Terai S, Fujisawa K, Takami T, Yamamoto N, Matsumoto T, Hirose Y, Murata Y, Yamasaki T, Sakaida I. Bortezomib induces tumor-specific cell death and growth inhibition in hepatocellular carcinoma and improves liver fibrosis. J. Gastroenterol. 2012;48:738–750. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
