

ALPK1 phosphorylates myosin IIA modulating TNF-α trafficking in gout flares
- PMID: 27169898
- PMCID: PMC4864424
- DOI: 10.1038/srep25740
ALPK1 phosphorylates myosin IIA modulating TNF-α trafficking in gout flares
Erratum in
-
Corrigendum: ALPK1 phosphorylates myosin IIA modulating TNF-α trafficking in gout flares.Sci Rep. 2016 Jun 10;6:27323. doi: 10.1038/srep27323. Sci Rep. 2016. PMID: 27283228 Free PMC article. No abstract available.
Abstract
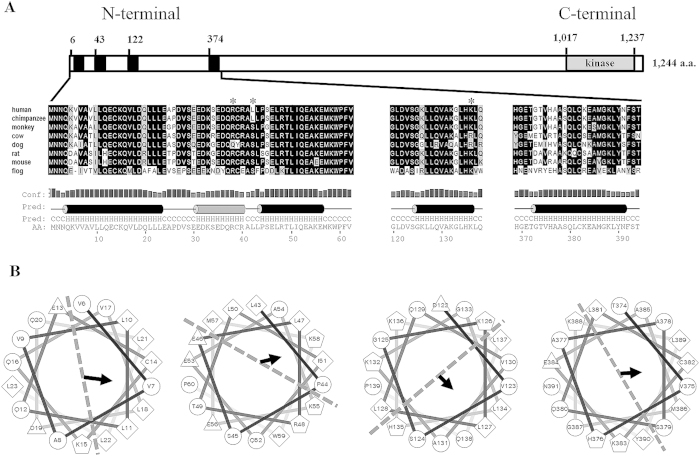
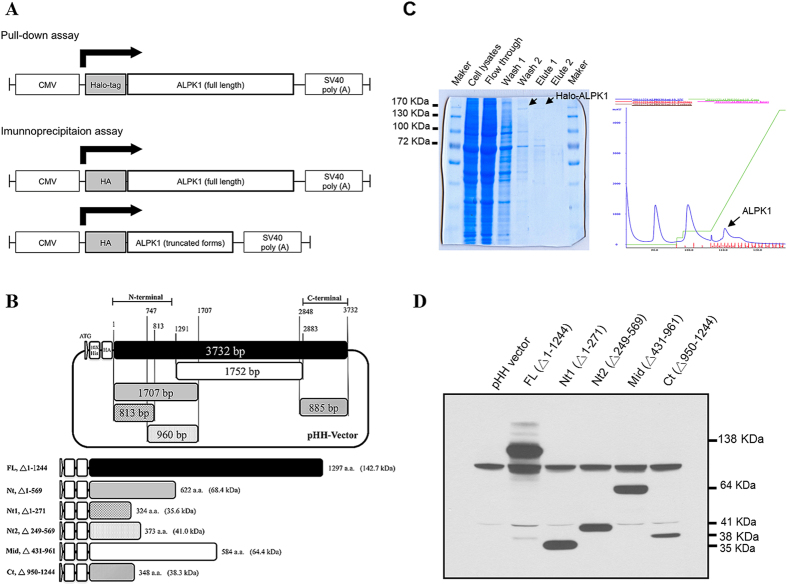
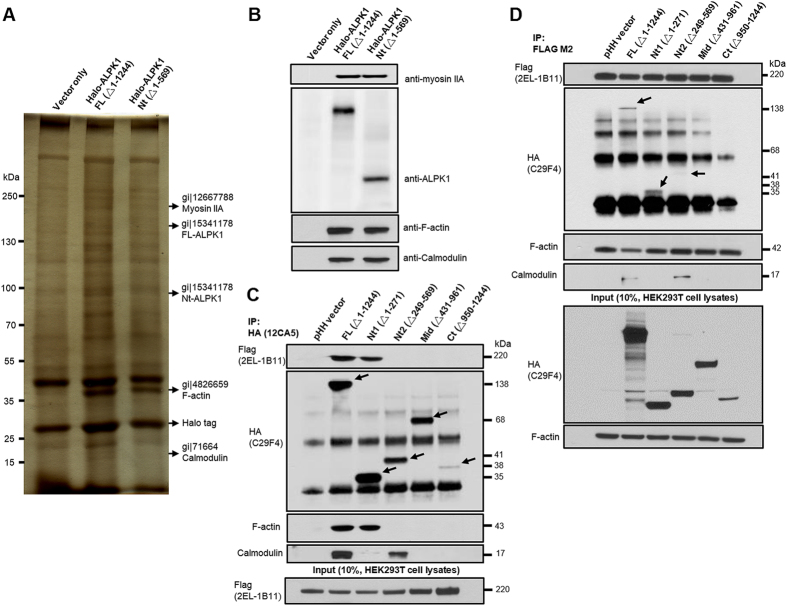
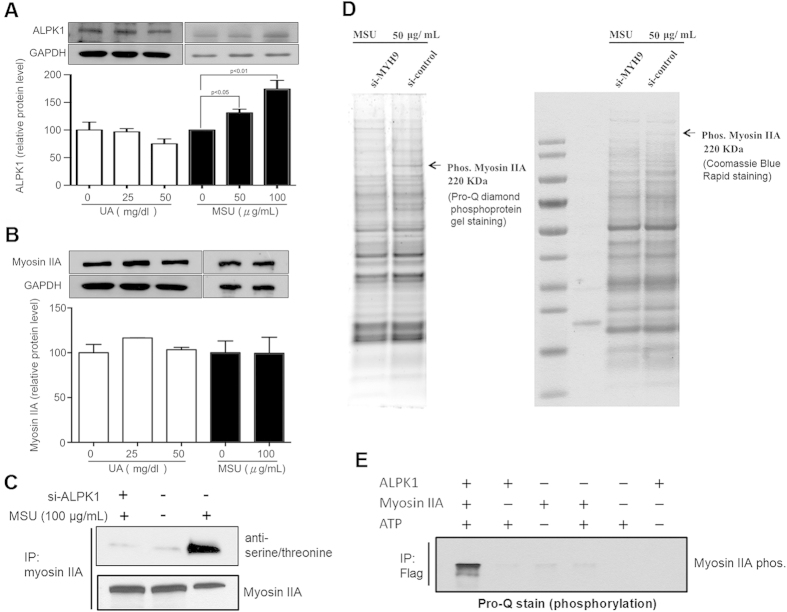
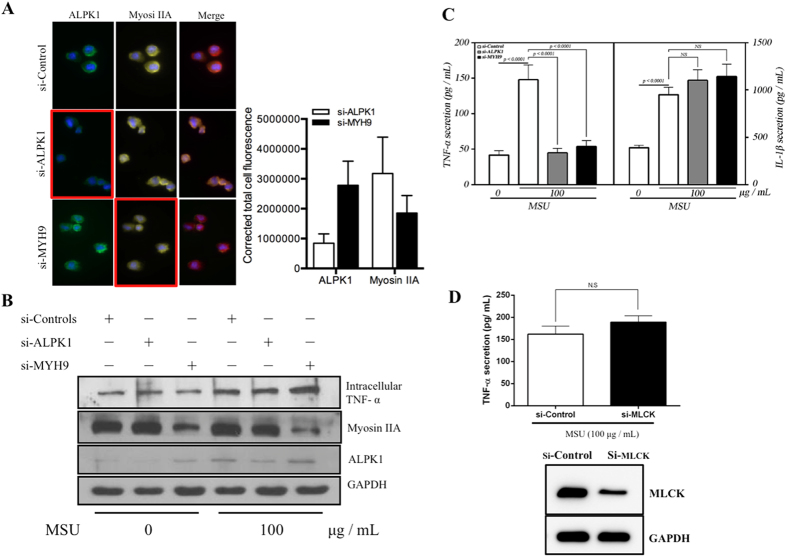
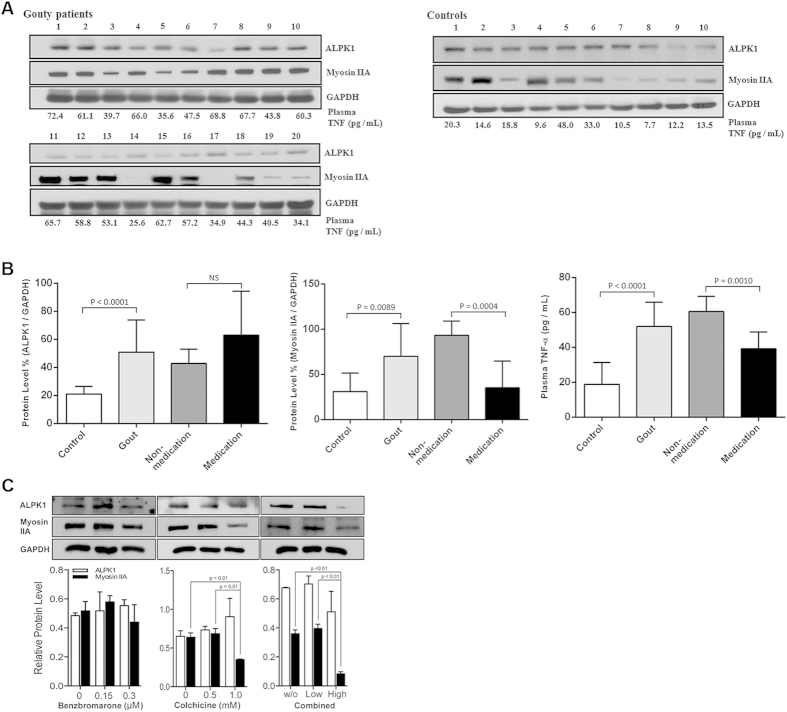
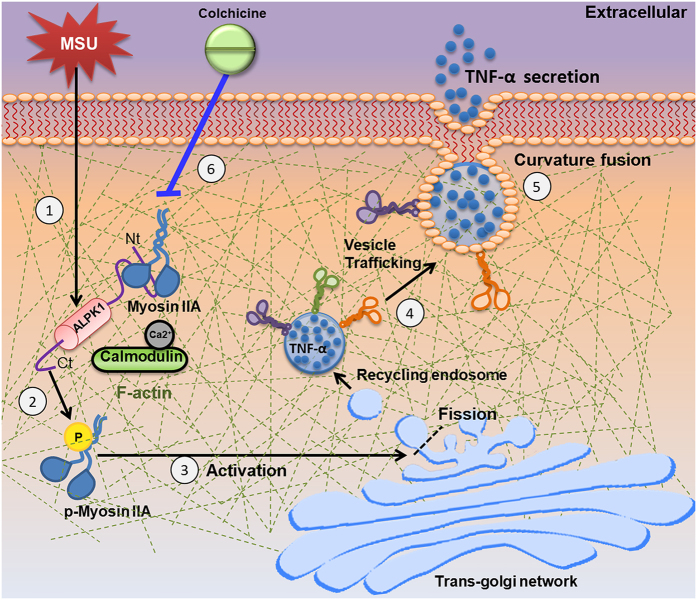
Gout is characterized by the monosodium urate monohydrate (MSU)-induced arthritis. Alpha kinase-1 (ALPK1) has shown to be associated with MSU-induced inflammation and gout. Here, we used bioinformatics, proteomics, cell models, and twenty in vitro human assays to clarify some of its role in the inflammatory response to MSU. We found myosin IIA to be a frequent interacting protein partner of ALPK1, binding to its N-terminal and forming a protein complex with calmodulin and F-actin, and that MSU-induced ALPK1 phosphorylated the myosin IIA. A knockdown of endogenous ALPK1 or myosin IIA significantly reduced the MSU-induced secretion of tumour necrosis factor (TNF)-α. Furthermore, all gouty patients expressed higher basal protein levels of ALPK1, myosin IIA, and plasma TNF-α, however those medicated with colchicine has shown reduced myosin IIA and TNF-α but not ALPK1. The findings suggest ALPK1 is a kinase that participates in the regulation of Golgi-derived TNF-α trafficking through myosin IIA phosphorylation in the inflammation of gout. This novel pathway could be blocked at the level of myosin by colchicine in gout treatment.
Figures
Similar articles
-
The importance of ALPK1 kinase functionality as a potential biomarker for inflammatory diseases.Mol Biol Rep. 2025 Jun 10;52(1):575. doi: 10.1007/s11033-025-10528-w. Mol Biol Rep. 2025. PMID: 40493324 Review.
-
Systematic Review of the Role of Alpha-Protein Kinase 1 in Cancer and Cancer-Related Inflammatory Diseases.Cancers (Basel). 2022 Sep 9;14(18):4390. doi: 10.3390/cancers14184390. Cancers (Basel). 2022. PMID: 36139553 Free PMC article. Review.
-
Lymphocyte α-kinase is a gout-susceptible gene involved in monosodium urate monohydrate-induced inflammatory responses.J Mol Med (Berl). 2011 Dec;89(12):1241-51. doi: 10.1007/s00109-011-0796-5. Epub 2011 Aug 7. J Mol Med (Berl). 2011. PMID: 21822924
-
Long noncoding RNA HAR1A regulates oral cancer progression through the alpha-kinase 1, bromodomain 7, and myosin IIA axis.J Mol Med (Berl). 2021 Sep;99(9):1323-1334. doi: 10.1007/s00109-021-02095-x. Epub 2021 Jun 7. J Mol Med (Berl). 2021. PMID: 34097087
-
URAT1 inhibition by ALPK1 is associated with uric acid homeostasis.Rheumatology (Oxford). 2017 Apr 1;56(4):654-659. doi: 10.1093/rheumatology/kew463. Rheumatology (Oxford). 2017. PMID: 28039413
Cited by
-
The importance of ALPK1 kinase functionality as a potential biomarker for inflammatory diseases.Mol Biol Rep. 2025 Jun 10;52(1):575. doi: 10.1007/s11033-025-10528-w. Mol Biol Rep. 2025. PMID: 40493324 Review.
-
Systematic Review of the Role of Alpha-Protein Kinase 1 in Cancer and Cancer-Related Inflammatory Diseases.Cancers (Basel). 2022 Sep 9;14(18):4390. doi: 10.3390/cancers14184390. Cancers (Basel). 2022. PMID: 36139553 Free PMC article. Review.
-
Alpha kinase 1 controls intestinal inflammation by suppressing the IL-12/Th1 axis.Nat Commun. 2018 Sep 18;9(1):3797. doi: 10.1038/s41467-018-06085-5. Nat Commun. 2018. PMID: 30228258 Free PMC article.
-
Variants of ALPK1 with ABCG2, SLC2A9, and SLC22A12 increased the positive predictive value for gout.J Hum Genet. 2018 Jan;63(1):63-70. doi: 10.1038/s10038-017-0368-9. Epub 2017 Nov 8. J Hum Genet. 2018. PMID: 29215084 Clinical Trial.
-
Gain-of-function mutations in ALPK1 cause an NF-κB-mediated autoinflammatory disease: functional assessment, clinical phenotyping and disease course of patients with ROSAH syndrome.Ann Rheum Dis. 2022 Oct;81(10):1453-1464. doi: 10.1136/annrheumdis-2022-222629. Epub 2022 Jul 22. Ann Rheum Dis. 2022. PMID: 35868845 Free PMC article.
References
-
- Wang S. J. et al. Lymphocyte alpha-kinase is a gout-susceptible gene involved in monosodium urate monohydrate-induced inflammatory responses. J Mol Med (Berl) 89, 1241–1251 (2011). - PubMed
-
- Vaillancourt J. P., Lyons C. & Cote G. P. Identification of two phosphorylated threonines in the tail region of Dictyostelium myosin II. J Biol Chem 263, 10082–10087 (1988). - PubMed
-
- Futey L. M., Medley Q. G., Cote G. P. & Egelhoff T. T. Structural analysis of myosin heavy chain kinase A from Dictyostelium. Evidence for a highly divergent protein kinase domain, an amino-terminal coiled-coil domain, and a domain homologous to the beta-subunit of heterotrimeric G proteins. J Biol Chem. 270, 523–529 (1995). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
