

Genome Sequence and Analysis of a Stress-Tolerant, Wild-Derived Strain of Saccharomyces cerevisiae Used in Biofuels Research
- PMID: 27172212
- PMCID: PMC4889671
- DOI: 10.1534/g3.116.029389
Genome Sequence and Analysis of a Stress-Tolerant, Wild-Derived Strain of Saccharomyces cerevisiae Used in Biofuels Research
Abstract
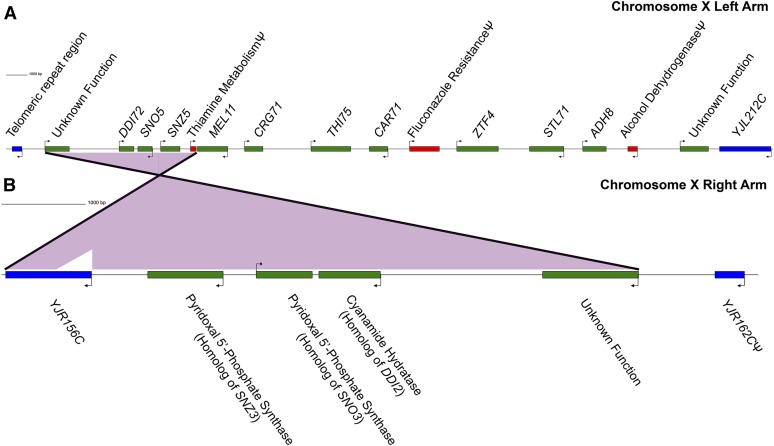
The genome sequences of more than 100 strains of the yeast Saccharomyces cerevisiae have been published. Unfortunately, most of these genome assemblies contain dozens to hundreds of gaps at repetitive sequences, including transposable elements, tRNAs, and subtelomeric regions, which is where novel genes generally reside. Relatively few strains have been chosen for genome sequencing based on their biofuel production potential, leaving an additional knowledge gap. Here, we describe the nearly complete genome sequence of GLBRCY22-3 (Y22-3), a strain of S. cerevisiae derived from the stress-tolerant wild strain NRRL YB-210 and subsequently engineered for xylose metabolism. After benchmarking several genome assembly approaches, we developed a pipeline to integrate Pacific Biosciences (PacBio) and Illumina sequencing data and achieved one of the highest quality genome assemblies for any S. cerevisiae strain. Specifically, the contig N50 is 693 kbp, and the sequences of most chromosomes, the mitochondrial genome, and the 2-micron plasmid are complete. Our annotation predicts 92 genes that are not present in the reference genome of the laboratory strain S288c, over 70% of which were expressed. We predicted functions for 43 of these genes, 28 of which were previously uncharacterized and unnamed. Remarkably, many of these genes are predicted to be involved in stress tolerance and carbon metabolism and are shared with a Brazilian bioethanol production strain, even though the strains differ dramatically at most genetic loci. The Y22-3 genome sequence provides an exceptionally high-quality resource for basic and applied research in bioenergy and genetics.
Keywords: Pacific Biosciences (PacBio); genome annotation; genome assembly; lignocellulosic hydrolysates; novel genes.
Copyright © 2016 McIlwain et al.
Figures





References
-
- Babrzadeh F., Jalili R., Wang C., Shokralla S., Pierce S., et al. , 2012. Whole-genome sequencing of the efficient industrial fuel-ethanol fermentative Saccharomyces cerevisiae strain CAT-1. Mol. Genet. Genomics 287: 485–494. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous