

Factor XI as a Therapeutic Target
- PMID: 27174099
- PMCID: PMC4919154
- DOI: 10.1161/ATVBAHA.116.306925
Factor XI as a Therapeutic Target
Abstract
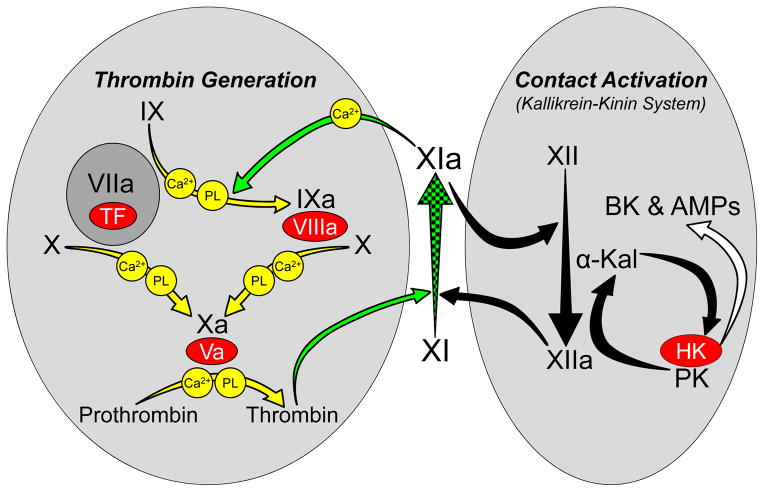
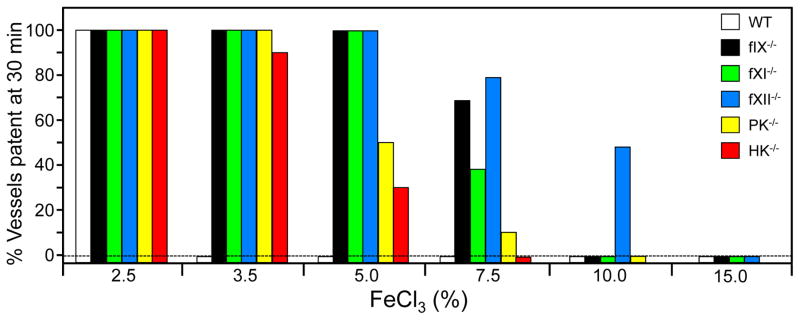
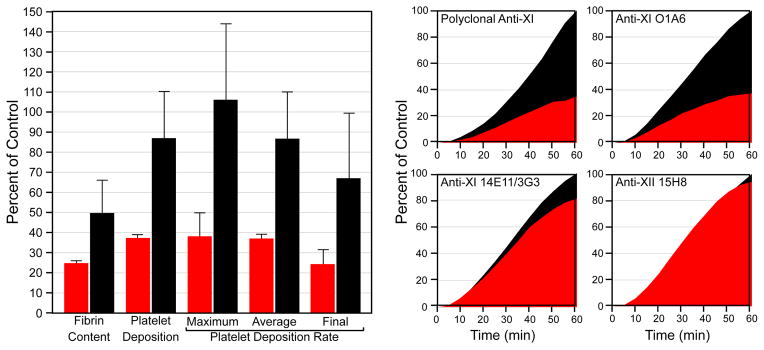
Factor XIa is a plasma serine protease that contributes to thrombin generation primarily through proteolytic activation of factor IX. Traditionally considered part of the intrinsic pathway of coagulation, several lines of evidence now suggest that factor XIa serves as an interface between the vitamin-K-dependent thrombin generation mechanism and the proinflammatory kallikrein-kinin system, allowing the 2 systems to influence each other. Work with animal models and results from epidemiological surveys of human populations support a role for factor XIa in thromboembolic disease. These data and the clinical observation that deficiency of factor XI, the zymogen of factor XIa, produces a relatively mild bleeding disorder suggest that drugs targeting factor XI or XIa could produce an antithrombotic effect while leaving hemostasis largely intact. Results of a recent trial comparing antisense-induced factor XI reduction to standard-dose low molecular-weight heparin as prophylaxis for venous thrombosis during knee replacement are encouraging in this regard. Here, we discuss recent findings on the biochemistry, physiology, and pathology of factor XI as they relate to thromboembolic disease.
Keywords: factor XI; factor XII; hemorrhage; thrombin; thrombosis.
© 2016 American Heart Association, Inc.
Figures
References
-
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood. 2008;111:4871–4879. - PubMed
-
- Hirsh J, O’Donnell M, Weitz JI. New Anticoagulants. Blood. 2005;105:453–463. - PubMed
-
- Yeh CH, Hogg K, Weitz JI. Overview of the new oral anticoagulants: opportunities and challenges. Arterioscler Thromb Vasc Biol. 2015;35:1056–1065. - PubMed
-
- Key NS. Epidemiologic and clinical data linking factors XI and XII to thrombosis. Hematology Am Soc Hematol Educ Program. 2014;2014:66–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
