

RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury
- PMID: 27177019
- PMCID: PMC5072432
- DOI: 10.1038/cdd.2016.46
RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury
Abstract
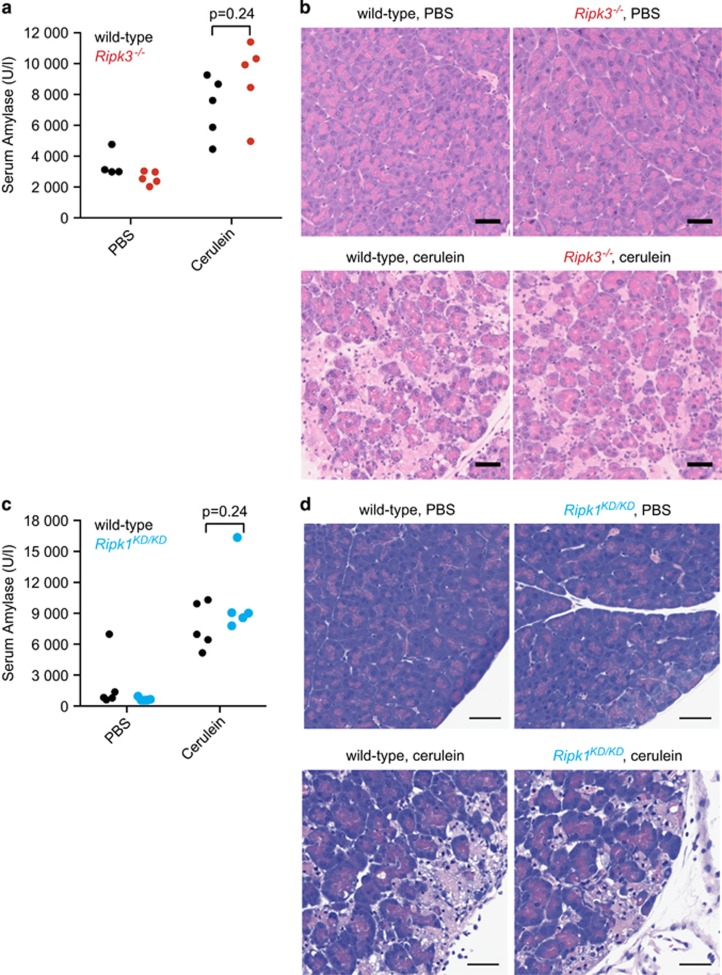
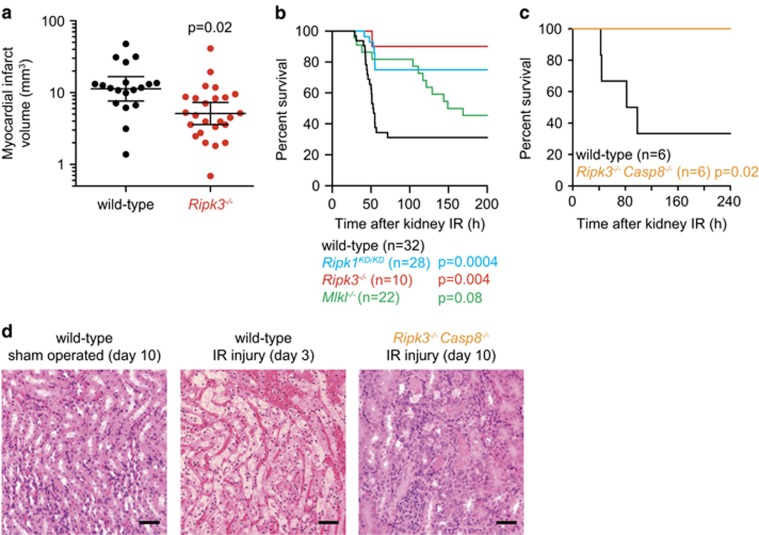
Necroptosis is a caspase-independent form of cell death that is triggered by activation of the receptor interacting serine/threonine kinase 3 (RIPK3) and phosphorylation of its pseudokinase substrate mixed lineage kinase-like (MLKL), which then translocates to membranes and promotes cell lysis. Activation of RIPK3 is regulated by the kinase RIPK1. Here we analyze the contribution of RIPK1, RIPK3, or MLKL to several mouse disease models. Loss of RIPK3 had no effect on lipopolysaccharide-induced sepsis, dextran sodium sulfate-induced colitis, cerulein-induced pancreatitis, hypoxia-induced cerebral edema, or the major cerebral artery occlusion stroke model. However, kidney ischemia-reperfusion injury, myocardial infarction, and systemic inflammation associated with A20 deficiency or high-dose tumor necrosis factor (TNF) were ameliorated by RIPK3 deficiency. Catalytically inactive RIPK1 was also beneficial in the kidney ischemia-reperfusion injury model, the high-dose TNF model, and in A20(-/-) mice. Interestingly, MLKL deficiency offered less protection in the kidney ischemia-reperfusion injury model and no benefit in A20(-/-) mice, consistent with necroptosis-independent functions for RIPK1 and RIPK3. Combined loss of RIPK3 (or MLKL) and caspase-8 largely prevented the cytokine storm, hypothermia, and morbidity induced by TNF, suggesting that the triggering event in this model is a combination of apoptosis and necroptosis. Tissue-specific RIPK3 deletion identified intestinal epithelial cells as the major target organ. Together these data emphasize that MLKL deficiency rather than RIPK1 inactivation or RIPK3 deficiency must be examined to implicate a role for necroptosis in disease.
Conflict of interest statement
All authors were employees of Genentech.
Figures





References
-
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336. - PubMed
-
- He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111. - PubMed
-
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–227. - PubMed
-
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013; 39: 443–453. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
