

Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability
- PMID: 27179618
- PMCID: PMC4868023
- DOI: 10.1186/s13023-016-0439-6
Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability
Abstract
Background: Mutations of TCF4, which encodes a basic helix-loop-helix transcription factor, cause Pitt-Hopkins syndrome (PTHS) via multiple genetic mechanisms. TCF4 is a complex locus expressing multiple transcripts by alternative splicing and use of multiple promoters. To address the relationship between mutation of these transcripts and phenotype, we report a three-generation family segregating mild intellectual disability with a chromosomal translocation disrupting TCF4.
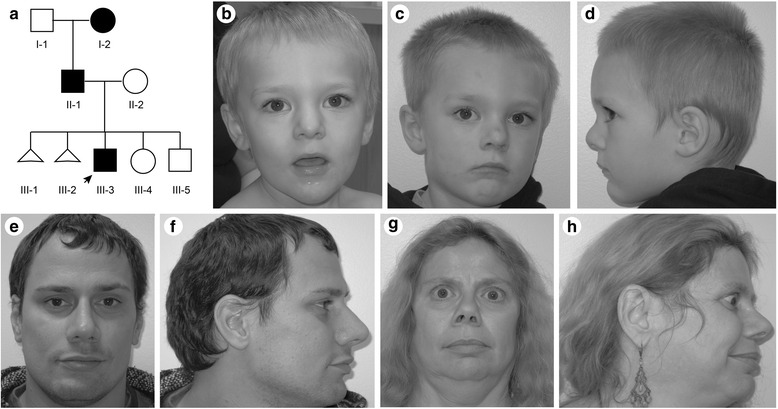
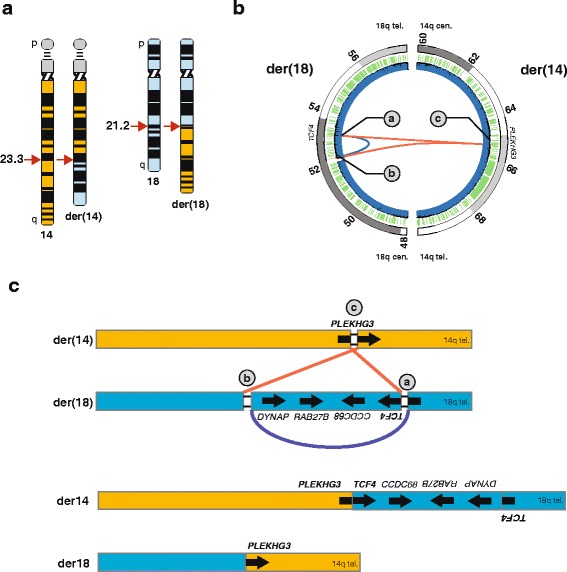
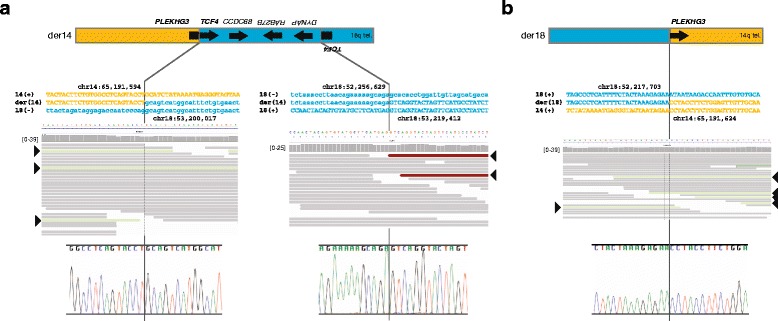
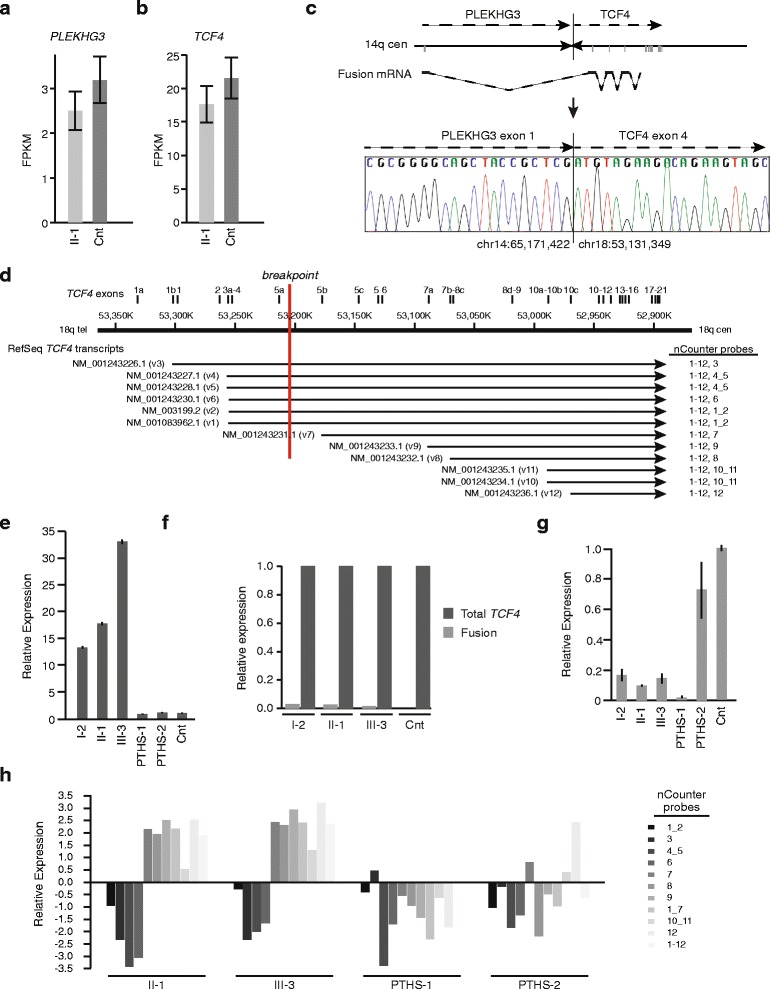
Results: Using whole genome sequencing, we detected a complex unbalanced karyotype disrupting TCF4 (46,XY,del(14)(q23.3q23.3)del(18)(q21.2q21.2)del(18)(q21.2q21.2)inv(18)(q21.2q21.2)t(14;18)(q23.3;q21.2)(14pter®14q23.3::18q21.2®18q21.2::18q21.1®18qter;18pter®18q21.2::14q23.3®14qter). Subsequent transcriptome sequencing, qRT-PCR and nCounter analyses revealed that cultured skin fibroblasts and peripheral blood had normal expression of genes along chromosomes 14 or 18 and no marked changes in expression of genes other than TCF4. Affected individuals had 12-33 fold higher mRNA levels of TCF4 than did unaffected controls or individuals with PTHS. Although the derivative chromosome generated a PLEKHG3-TCF4 fusion transcript, the increased levels of TCF4 mRNA arose from transcript variants originating distal to the translocation breakpoint, not from the fusion transcript.
Conclusions: Although validation in additional patients is required, our findings suggest that the dysmorphic features and severe intellectual disability characteristic of PTHS are partially rescued by overexpression of those short TCF4 transcripts encoding a nuclear localization signal, a transcription activation domain, and the basic helix-loop-helix domain.
Keywords: Gene expression; Intellectual disability; Pitt-Hopkins syndrome; Promoter utilization; RNAseq; TCF4; Transcriptome; Translocation.
Figures
Similar articles
-
Partial deletion of TCF4 in three generation family with non-syndromic intellectual disability, without features of Pitt-Hopkins syndrome.Eur J Med Genet. 2016 Jun;59(6-7):310-4. doi: 10.1016/j.ejmg.2016.04.003. Epub 2016 Apr 28. Eur J Med Genet. 2016. PMID: 27132474
-
The Pitt-Hopkins syndrome: report of 16 new patients and clinical diagnostic criteria.Am J Med Genet A. 2011 Jul;155A(7):1536-45. doi: 10.1002/ajmg.a.34070. Epub 2011 Jun 10. Am J Med Genet A. 2011. PMID: 21671391
-
Disruption and deletion of the proximal part of TCF4 are associated with mild intellectual disability: About three new patients.Eur J Med Genet. 2022 Apr;65(4):104458. doi: 10.1016/j.ejmg.2022.104458. Epub 2022 Feb 18. Eur J Med Genet. 2022. PMID: 35189377
-
The emerging roles of TCF4 in disease and development.Trends Mol Med. 2014 Jun;20(6):322-31. doi: 10.1016/j.molmed.2014.01.010. Epub 2014 Mar 1. Trends Mol Med. 2014. PMID: 24594265 Review.
-
Pitt-Hopkins Syndrome: intellectual disability due to loss of TCF4-regulated gene transcription.Exp Mol Med. 2013 May 3;45(5):e21. doi: 10.1038/emm.2013.32. Exp Mol Med. 2013. PMID: 23640545 Free PMC article. Review.
Cited by
-
WNT/β-Catenin Pathway and Epigenetic Mechanisms Regulate the Pitt-Hopkins Syndrome and Schizophrenia Risk Gene TCF4.Mol Neuropsychiatry. 2017 Jul;3(1):53-71. doi: 10.1159/000475666. Epub 2017 Jul 14. Mol Neuropsychiatry. 2017. PMID: 28879201 Free PMC article.
-
Deciphering the role of precursor miR-12136 and miR-8485 in the progression of intellectual disability (ID).IBRO Neurosci Rep. 2022 Oct 21;13:393-401. doi: 10.1016/j.ibneur.2022.10.005. eCollection 2022 Dec. IBRO Neurosci Rep. 2022. PMID: 36345471 Free PMC article.
-
Region and Cell Type Distribution of TCF4 in the Postnatal Mouse Brain.Front Neuroanat. 2020 Jul 17;14:42. doi: 10.3389/fnana.2020.00042. eCollection 2020. Front Neuroanat. 2020. PMID: 32765228 Free PMC article.
-
Functional consequences of TCF4 missense substitutions associated with Pitt-Hopkins syndrome, mild intellectual disability, and schizophrenia.J Biol Chem. 2021 Dec;297(6):101381. doi: 10.1016/j.jbc.2021.101381. Epub 2021 Nov 6. J Biol Chem. 2021. PMID: 34748727 Free PMC article.
-
Pitt-Hopkins Syndrome: Clinical and Molecular Findings of a 5-Year-Old Patient.Genes (Basel). 2020 May 28;11(6):596. doi: 10.3390/genes11060596. Genes (Basel). 2020. PMID: 32481733 Free PMC article.
References
-
- Luckasson R, et al. Mental Retardation: Definition, Classification, and Systems of Supports 10. Washington, DC: The American Association on Intellecutal and Developmental Disabilitites; 2002.
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
