

Transition from inflammation to proliferation: a critical step during wound healing
- PMID: 27180275
- PMCID: PMC5021733
- DOI: 10.1007/s00018-016-2268-0
Transition from inflammation to proliferation: a critical step during wound healing
Abstract
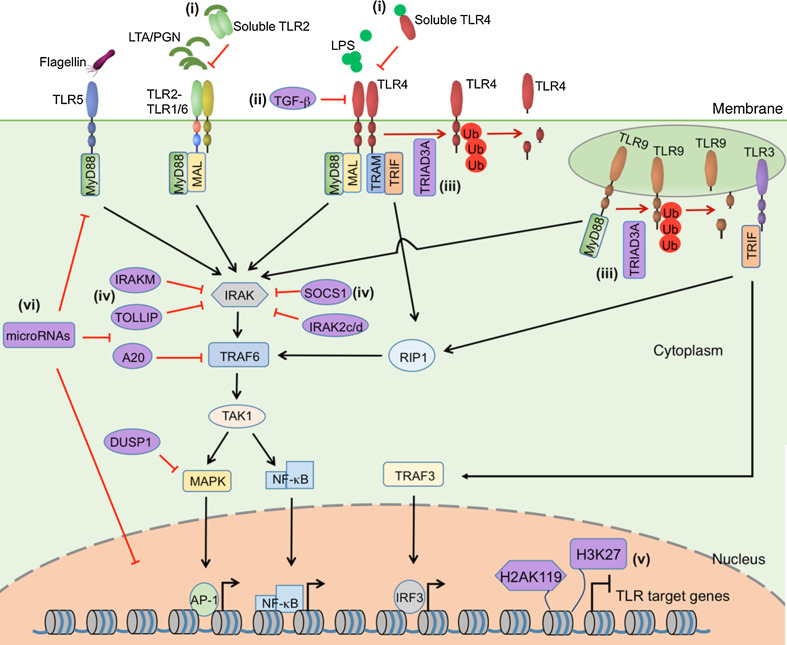
The ability to rapidly restore the integrity of a broken skin barrier is critical and is the ultimate goal of therapies for hard-to-heal-ulcers. Unfortunately effective treatments to enhance healing and reduce scarring are still lacking. A deeper understanding of the physiology of normal repair and of the pathology of delayed healing is a prerequisite for the development of more effective therapeutic interventions. Transition from the inflammatory to the proliferative phase is a key step during healing and accumulating evidence associates a compromised transition with wound healing disorders. Thus, targeting factors that impact this phase transition may offer a rationale for therapeutic development. This review summarizes mechanisms regulating the inflammation-proliferation transition at cellular and molecular levels. We propose that identification of such mechanisms will reveal promising targets for development of more effective therapies.
Keywords: Bioactive lipid mediator; Fibroblast; Macrophage; MicroRNA; Reactive oxygen species; Toll-like receptor; Transcription factor.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
