

Potentiation of Glucose-stimulated Insulin Secretion by the GPR40-PLC-TRPC Pathway in Pancreatic β-Cells
- PMID: 27180622
- PMCID: PMC4867641
- DOI: 10.1038/srep25912
Potentiation of Glucose-stimulated Insulin Secretion by the GPR40-PLC-TRPC Pathway in Pancreatic β-Cells
Abstract
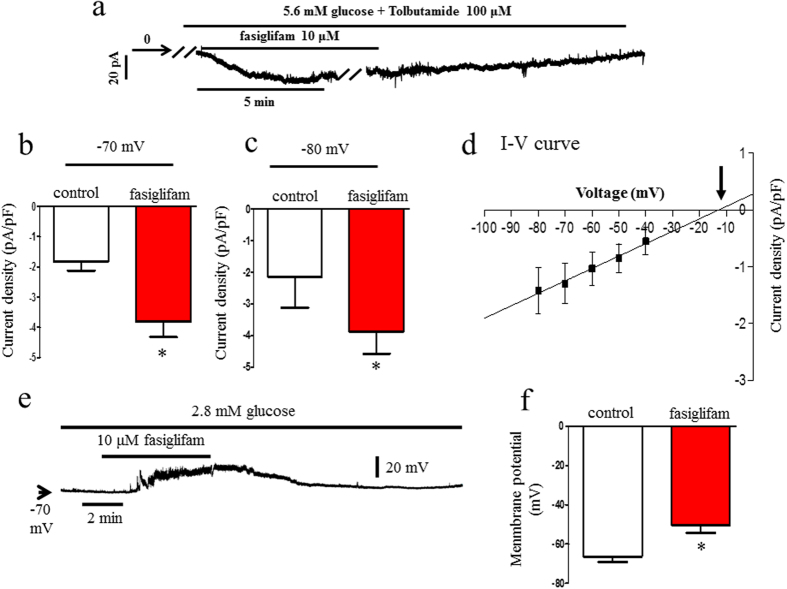
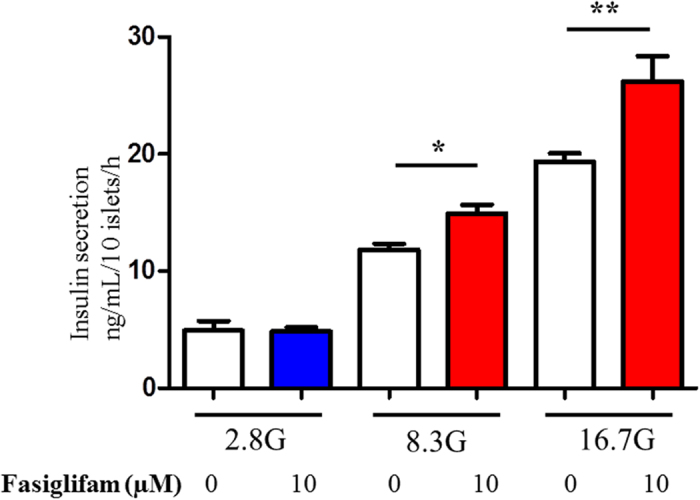
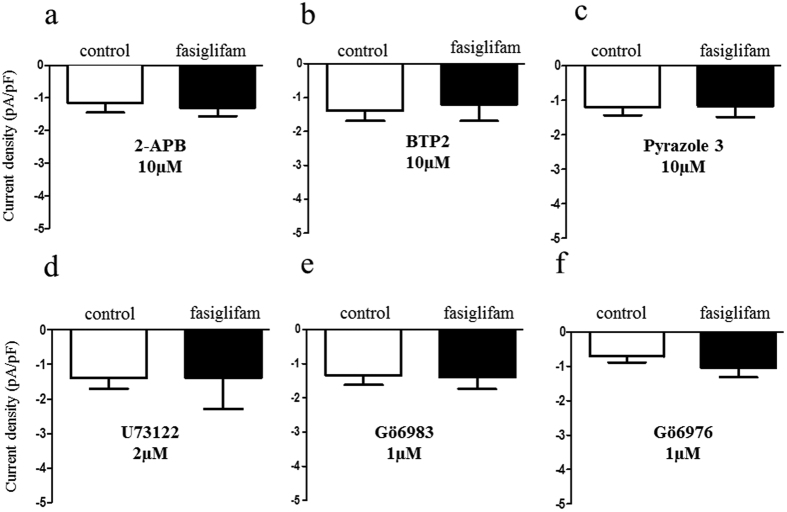
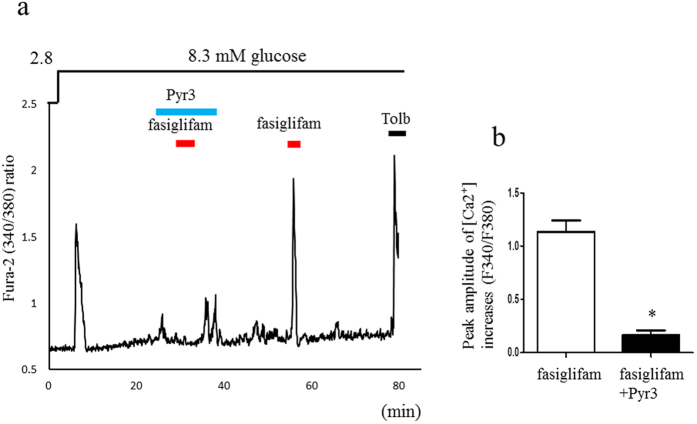
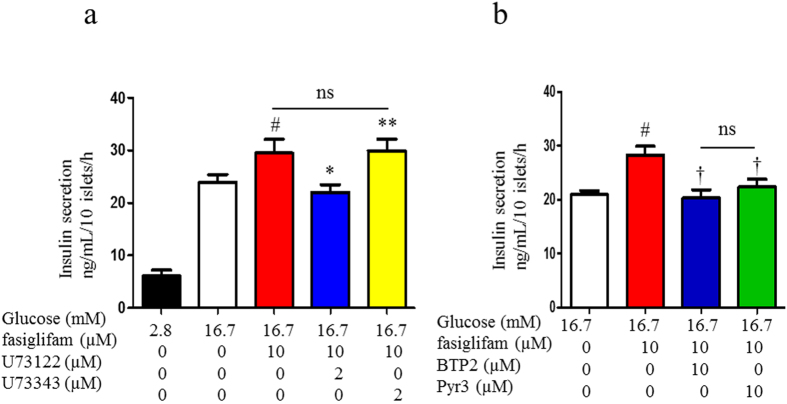
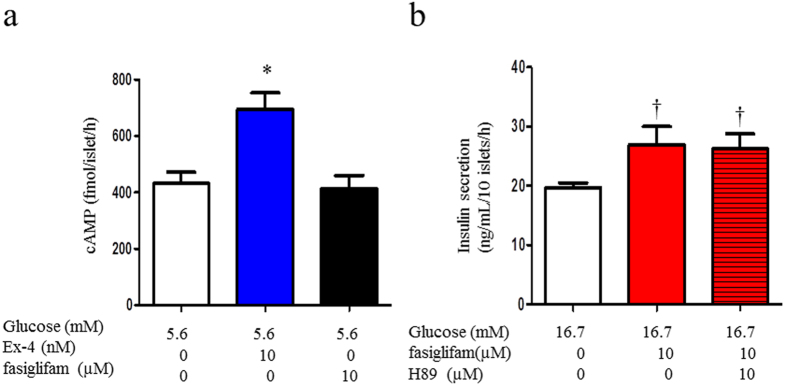
G protein-coupled receptors (GPCRs) are expressed in pancreatic beta-cells. G protein-coupled receptor 40 (GPR40) contributes to medium- or long-chain fatty acid-induced amplification of glucose-stimulated insulin secretion (GSIS), and GPR40 agonists are promising therapeutic targets in type 2 diabetes. Recently, we demonstrated that glucagon-like peptide 1, a ligand of pancreatic GPCR, activates a class of nonselective cation channels (NSCCs) and enhances GSIS. The aim of the current study was to determine whether the GPR40 signal interacts with NSCCs. A GPR40 agonist (fasiglifam) potentiated GSIS at 8.3 and 16.7 mM glucose but not 2.8 mM glucose. The NSCC current was activated by fasiglifam at 5.6 mM glucose with 100 μM tolbutamide (-70 mV), and this activation was prevented by the presence of pyrazole-3 (transient receptor potential canonical; a TRPC3 channel blocker). Inhibitors of phospholipase C or protein kinase C (PKC) inhibited the increases in GSIS and the NSCC current induced by GPR40 stimulation. The present study demonstrates a novel mechanism for the regulation of insulin secretion by GPR40 agonist in pancreatic beta-cells. The stimulation of the GPR40-PLC/PKC-TRPC3 channel pathway potentiates GSIS by the depolarization of the plasma membrane in pancreatic beta-cell.
Figures
Similar articles
-
Protective role of Pollen Typhae total flavone against the palmitic acid-induced impairment of glucose-stimulated insulin secretion involving GPR40 signaling in INS-1 cells.Int J Mol Med. 2017 Sep;40(3):922-930. doi: 10.3892/ijmm.2017.3070. Epub 2017 Jul 18. Int J Mol Med. 2017. PMID: 28731171
-
Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel and link to insulin release.Am J Physiol Endocrinol Metab. 2005 Oct;289(4):E670-7. doi: 10.1152/ajpendo.00035.2005. Epub 2005 May 24. Am J Physiol Endocrinol Metab. 2005. PMID: 15914509
-
The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets.J Pharmacol Exp Ther. 2012 Feb;340(2):483-9. doi: 10.1124/jpet.111.187708. Epub 2011 Nov 21. J Pharmacol Exp Ther. 2012. PMID: 22106100
-
GPR40: a therapeutic target for mediating insulin secretion (review).Int J Mol Med. 2012 Dec;30(6):1261-6. doi: 10.3892/ijmm.2012.1142. Epub 2012 Sep 26. Int J Mol Med. 2012. PMID: 23023155 Review.
-
Glucose and GTP-binding protein-coupled receptor cooperatively regulate transient receptor potential-channels to stimulate insulin secretion [Review].Endocr J. 2016 Oct 29;63(10):867-876. doi: 10.1507/endocrj.EJ16-0262. Epub 2016 Jul 17. Endocr J. 2016. PMID: 27321586 Review.
Cited by
-
Transient Receptor Potential Canonical (TRPC) Channels: Then and Now.Cells. 2020 Aug 28;9(9):1983. doi: 10.3390/cells9091983. Cells. 2020. PMID: 32872338 Free PMC article. Review.
-
Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men.Physiol Rev. 2018 Jan 1;98(1):117-214. doi: 10.1152/physrev.00008.2017. Physiol Rev. 2018. PMID: 29212789 Free PMC article. Review.
-
Free fatty acid receptors in the endocrine regulation of glucose metabolism: Insight from gastrointestinal-pancreatic-adipose interactions.Front Endocrinol (Lausanne). 2022 Sep 28;13:956277. doi: 10.3389/fendo.2022.956277. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36246919 Free PMC article. Review.
-
G-protein-coupled receptor 40 agonist GW9508 potentiates glucose-stimulated insulin secretion through activation of protein kinase Cα and ε in INS-1 cells.PLoS One. 2019 Sep 9;14(9):e0222179. doi: 10.1371/journal.pone.0222179. eCollection 2019. PLoS One. 2019. PMID: 31498851 Free PMC article.
-
Beta-Cell Ion Channels and Their Role in Regulating Insulin Secretion.Compr Physiol. 2021 Oct 12;11(4):1-21. doi: 10.1002/cphy.c210004. Compr Physiol. 2021. PMID: 34636409 Free PMC article. Review.
References
-
- Ahren B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov 8, 369–385 (2009). - PubMed
-
- Tomita T. et al. Expression of the gene for a membrane-bound fatty acid receptor in the pancreas and islet cell tumours in humans: evidence for GPR40 expression in pancreatic beta cells and implications for insulin secretion. Diabetologia 49, 962–968 (2006). - PubMed
-
- Itoh Y. et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422, 173–176 (2003). - PubMed
-
- Araki T., Hirayama M., Hiroi S. & Kaku K. GPR40-induced insulin secretion by the novel agonist TAK-875: first clinical findings in patients with type 2 diabetes. Diabetes Obes Metab 14, 271–278 (2012). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
