

ML418: The First Selective, Sub-Micromolar Pore Blocker of Kir7.1 Potassium Channels
- PMID: 27184474
- PMCID: PMC5131535
- DOI: 10.1021/acschemneuro.6b00111
ML418: The First Selective, Sub-Micromolar Pore Blocker of Kir7.1 Potassium Channels
Abstract
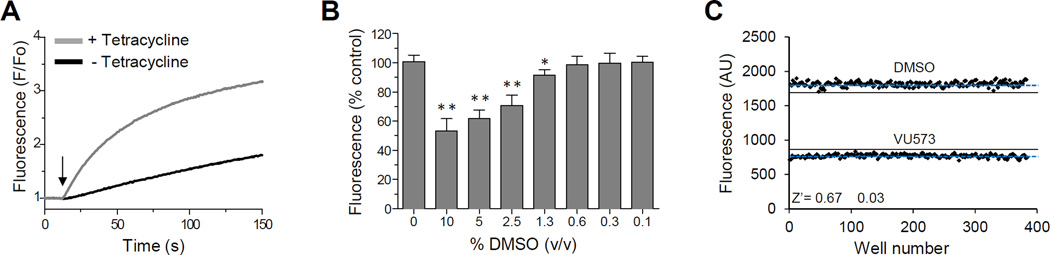
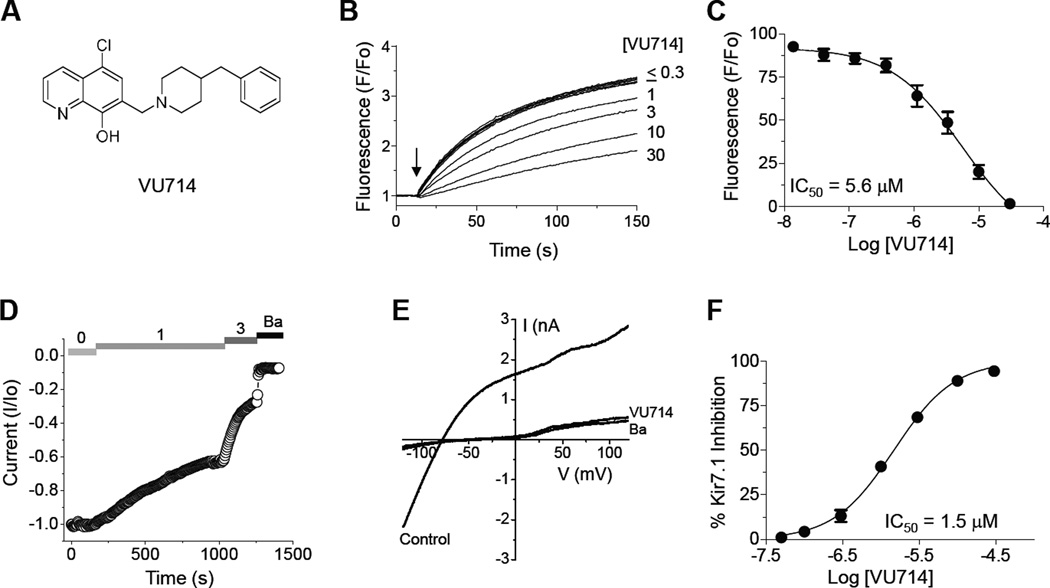
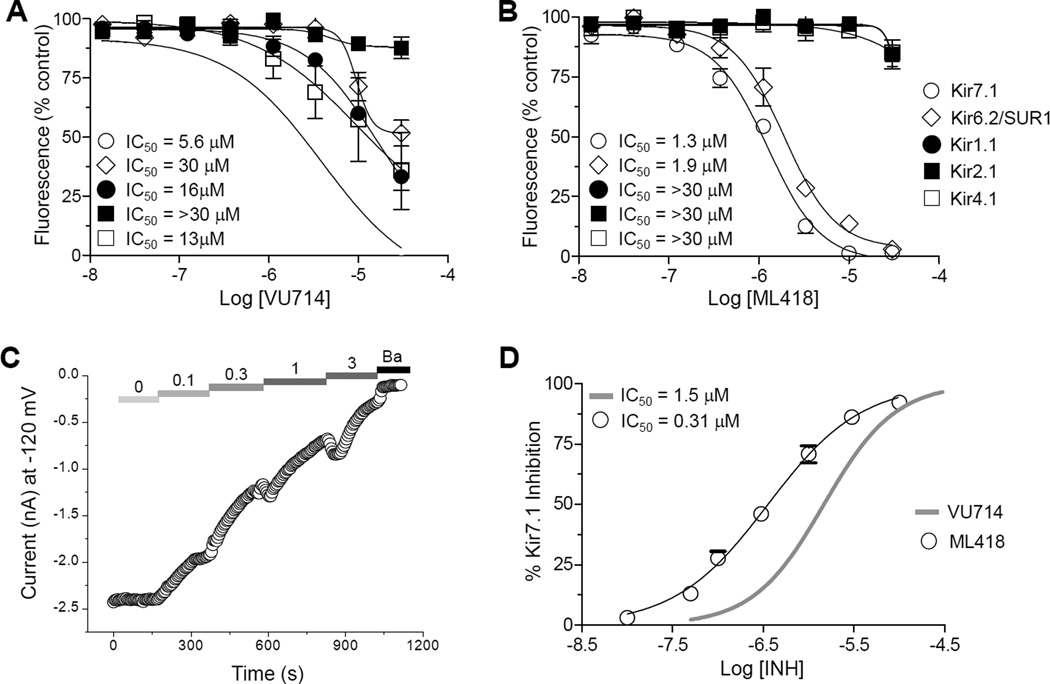
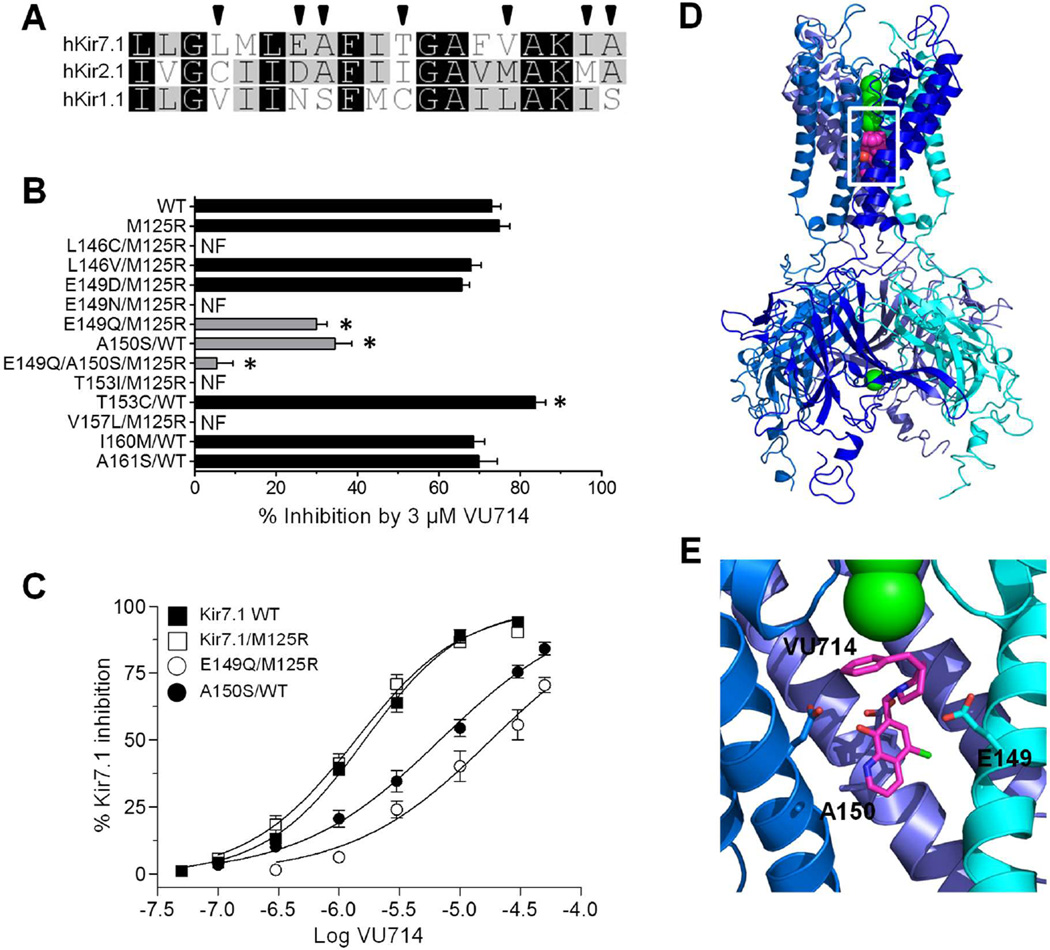
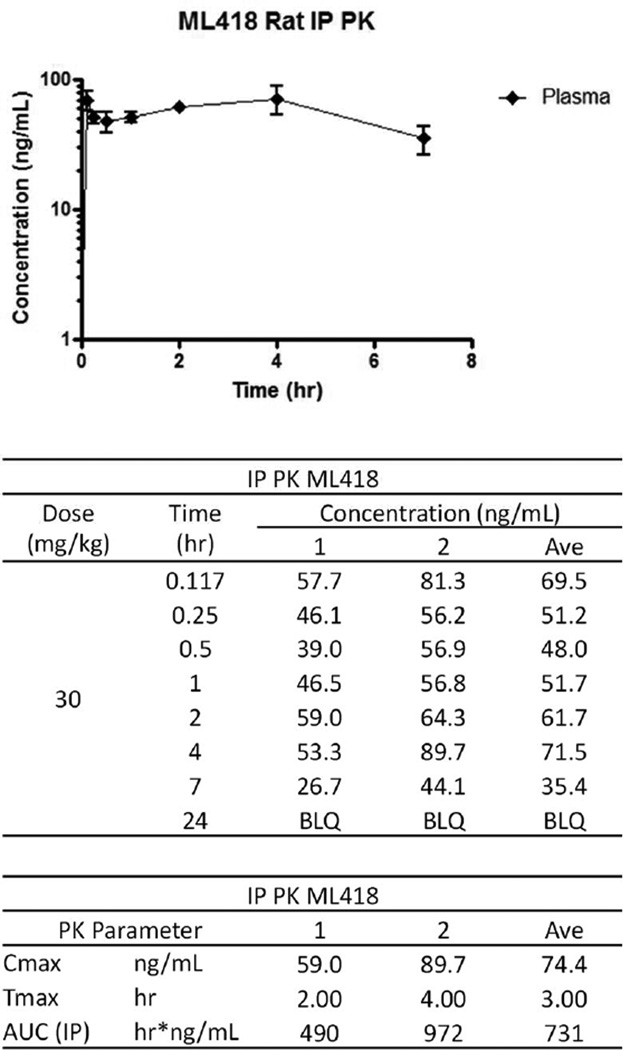
The inward rectifier potassium (Kir) channel Kir7.1 (KCNJ13) has recently emerged as a key regulator of melanocortin signaling in the brain, electrolyte homeostasis in the eye, and uterine muscle contractility during pregnancy. The pharmacological tools available for exploring the physiology and therapeutic potential of Kir7.1 have been limited to relatively weak and nonselective small-molecule inhibitors. Here, we report the discovery in a fluorescence-based high-throughput screen of a novel Kir7.1 channel inhibitor, VU714. Site-directed mutagenesis of pore-lining amino acid residues identified glutamate 149 and alanine 150 as essential determinants of VU714 activity. Lead optimization with medicinal chemistry generated ML418, which exhibits sub-micromolar activity (IC50 = 310 nM) and superior selectivity over other Kir channels (at least 17-fold selective over Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, and Kir4.1) except for Kir6.2/SUR1 (equally potent). Evaluation in the EuroFins Lead Profiling panel of 64 GPCRs, ion-channels, and transporters for off-target activity of ML418 revealed a relatively clean ancillary pharmacology. While ML418 exhibited low CLHEP in human microsomes which could be modulated with lipophilicity adjustments, it showed high CLHEP in rat microsomes regardless of lipophilicity. A subsequent in vivo PK study of ML418 by intraperitoneal (IP) administration (30 mg/kg dosage) revealed a suitable PK profile (Cmax = 0.20 μM and Tmax = 3 h) and favorable CNS distribution (mouse brain/plasma Kp of 10.9 to support in vivo studies. ML418, which represents the current state-of-the-art in Kir7.1 inhibitors, should be useful for exploring the physiology of Kir7.1 in vitro and in vivo.
Keywords: KCNJ13; comparative modeling; electrophysiology; melanocortin signaling; myometrium; thallium flux.
Figures
Similar articles
-
Pore Polarity and Charge Determine Differential Block of Kir1.1 and Kir7.1 Potassium Channels by Small-Molecule Inhibitor VU590.Mol Pharmacol. 2017 Sep;92(3):338-346. doi: 10.1124/mol.117.108472. Epub 2017 Jun 15. Mol Pharmacol. 2017. PMID: 28619748 Free PMC article.
-
Next-generation inward rectifier potassium channel modulators: discovery and molecular pharmacology.Am J Physiol Cell Physiol. 2021 Jun 1;320(6):C1125-C1140. doi: 10.1152/ajpcell.00548.2020. Epub 2021 Apr 7. Am J Physiol Cell Physiol. 2021. PMID: 33826405 Free PMC article. Review.
-
Discovery, Characterization, and Effects on Renal Fluid and Electrolyte Excretion of the Kir4.1 Potassium Channel Pore Blocker, VU0134992.Mol Pharmacol. 2018 Aug;94(2):926-937. doi: 10.1124/mol.118.112359. Epub 2018 Jun 12. Mol Pharmacol. 2018. PMID: 29895592 Free PMC article.
-
VU6036720: The First Potent and Selective In Vitro Inhibitor of Heteromeric Kir4.1/5.1 Inward Rectifier Potassium Channels.Mol Pharmacol. 2022 May;101(5):357-370. doi: 10.1124/molpharm.121.000464. Epub 2022 Mar 3. Mol Pharmacol. 2022. PMID: 35246480 Free PMC article.
-
Focus on Kir7.1: physiology and channelopathy.Channels (Austin). 2014;8(6):488-95. doi: 10.4161/19336950.2014.959809. Channels (Austin). 2014. PMID: 25558901 Free PMC article. Review.
Cited by
-
Automated Patch Clamp Recordings of GPCR-Gated Ion Channels: Targeting the MC4-R/Kir7.1 Potassium Channel Complex.Methods Mol Biol. 2024;2796:229-248. doi: 10.1007/978-1-0716-3818-7_14. Methods Mol Biol. 2024. PMID: 38856905
-
Pore Polarity and Charge Determine Differential Block of Kir1.1 and Kir7.1 Potassium Channels by Small-Molecule Inhibitor VU590.Mol Pharmacol. 2017 Sep;92(3):338-346. doi: 10.1124/mol.117.108472. Epub 2017 Jun 15. Mol Pharmacol. 2017. PMID: 28619748 Free PMC article.
-
Kir7.1 is the physiological target for hormones and steroids that regulate uteroplacental function.Sci Adv. 2025 Mar 7;11(10):eadr5086. doi: 10.1126/sciadv.adr5086. Epub 2025 Mar 5. Sci Adv. 2025. PMID: 40043131 Free PMC article.
-
Potassium channels in intestinal epithelial cells and their pharmacological modulation: a systematic review.Am J Physiol Cell Physiol. 2021 Apr 1;320(4):C520-C546. doi: 10.1152/ajpcell.00393.2020. Epub 2020 Dec 16. Am J Physiol Cell Physiol. 2021. PMID: 33326312 Free PMC article.
-
Next-generation inward rectifier potassium channel modulators: discovery and molecular pharmacology.Am J Physiol Cell Physiol. 2021 Jun 1;320(6):C1125-C1140. doi: 10.1152/ajpcell.00548.2020. Epub 2021 Apr 7. Am J Physiol Cell Physiol. 2021. PMID: 33826405 Free PMC article. Review.
References
-
- Ookata K, Tojo A, Suzuki Y, Nakamura N, Kimura K, Wilcox CS, Hirose S. Localization of inward rectifier potassium channel Kir7.1 in the basolateral membrane of distal nephron and collecting duct. J Am Soc Nephrol. 2000;11(11):1987–1994. - PubMed
-
- Nakamura N, Suzuki Y, Sakuta H, Ookata K, Kawahara K, Hirose S. Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+ K+-ATPase. Biochem J. 1999;342(Pt 2):329–336. - PMC - PubMed
-
- Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. A novel inward rectifier K+ channel with unique pore properties. Neuron. 1998;20(5):995–1005. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
