

Coming of age: ten years of next-generation sequencing technologies
- PMID: 27184599
- PMCID: PMC10373632
- DOI: 10.1038/nrg.2016.49
Coming of age: ten years of next-generation sequencing technologies
Abstract
Since the completion of the human genome project in 2003, extraordinary progress has been made in genome sequencing technologies, which has led to a decreased cost per megabase and an increase in the number and diversity of sequenced genomes. An astonishing complexity of genome architecture has been revealed, bringing these sequencing technologies to even greater advancements. Some approaches maximize the number of bases sequenced in the least amount of time, generating a wealth of data that can be used to understand increasingly complex phenotypes. Alternatively, other approaches now aim to sequence longer contiguous pieces of DNA, which are essential for resolving structurally complex regions. These and other strategies are providing researchers and clinicians a variety of tools to probe genomes in greater depth, leading to an enhanced understanding of how genome sequence variants underlie phenotype and disease.
Conflict of interest statement
Competing interests statement
The authors declare competing interests: see
Figures
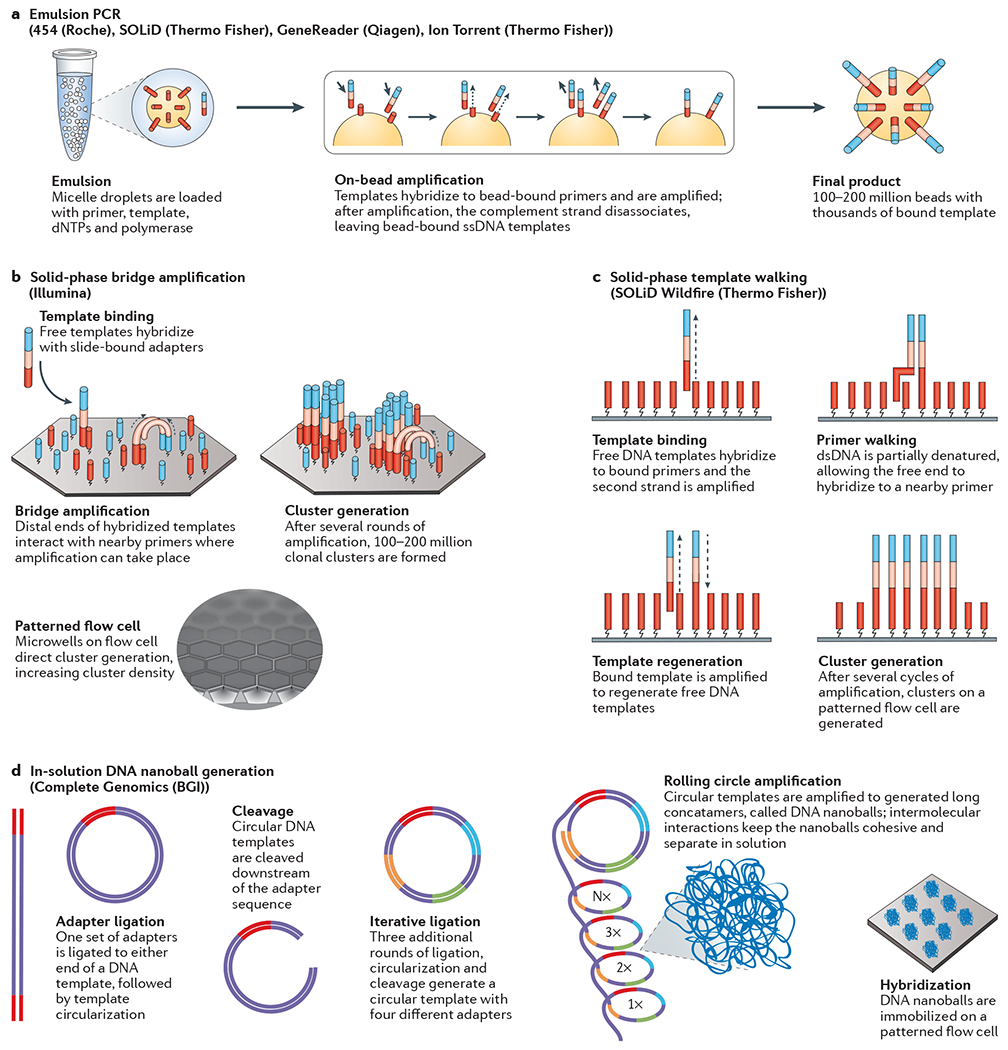
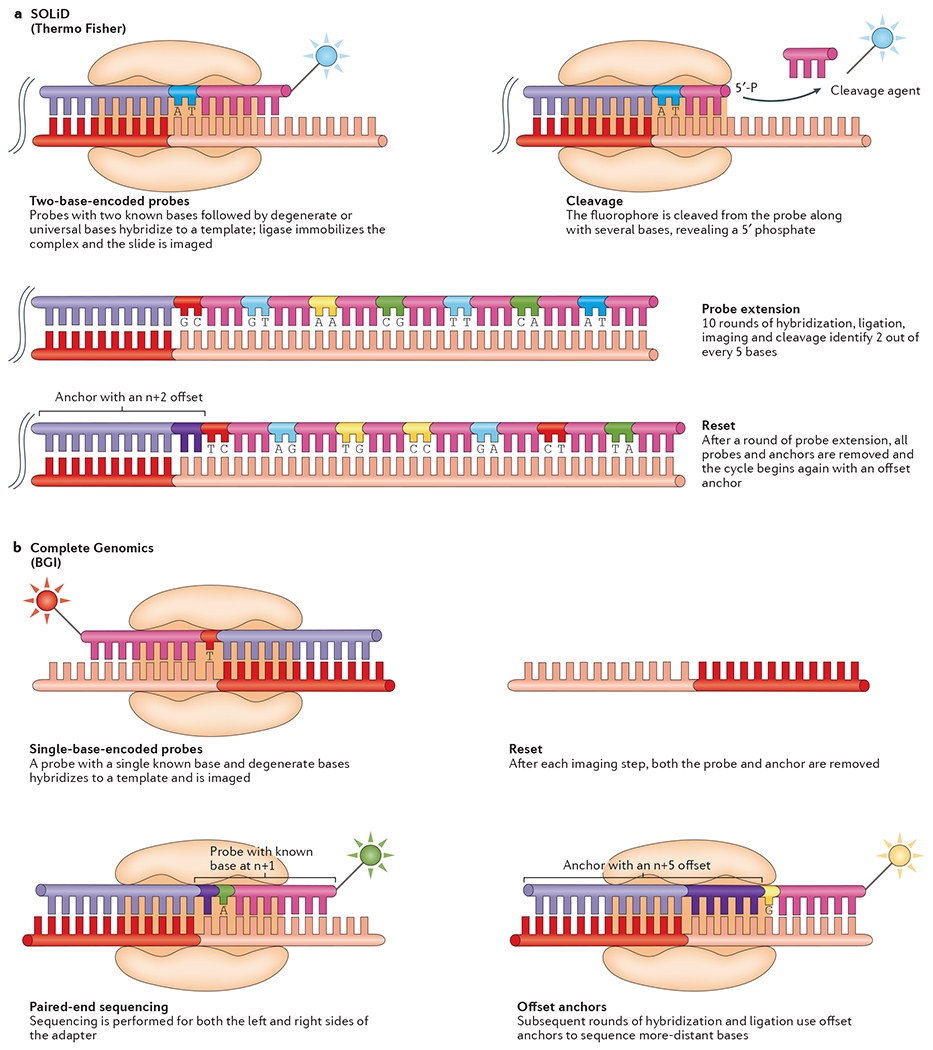
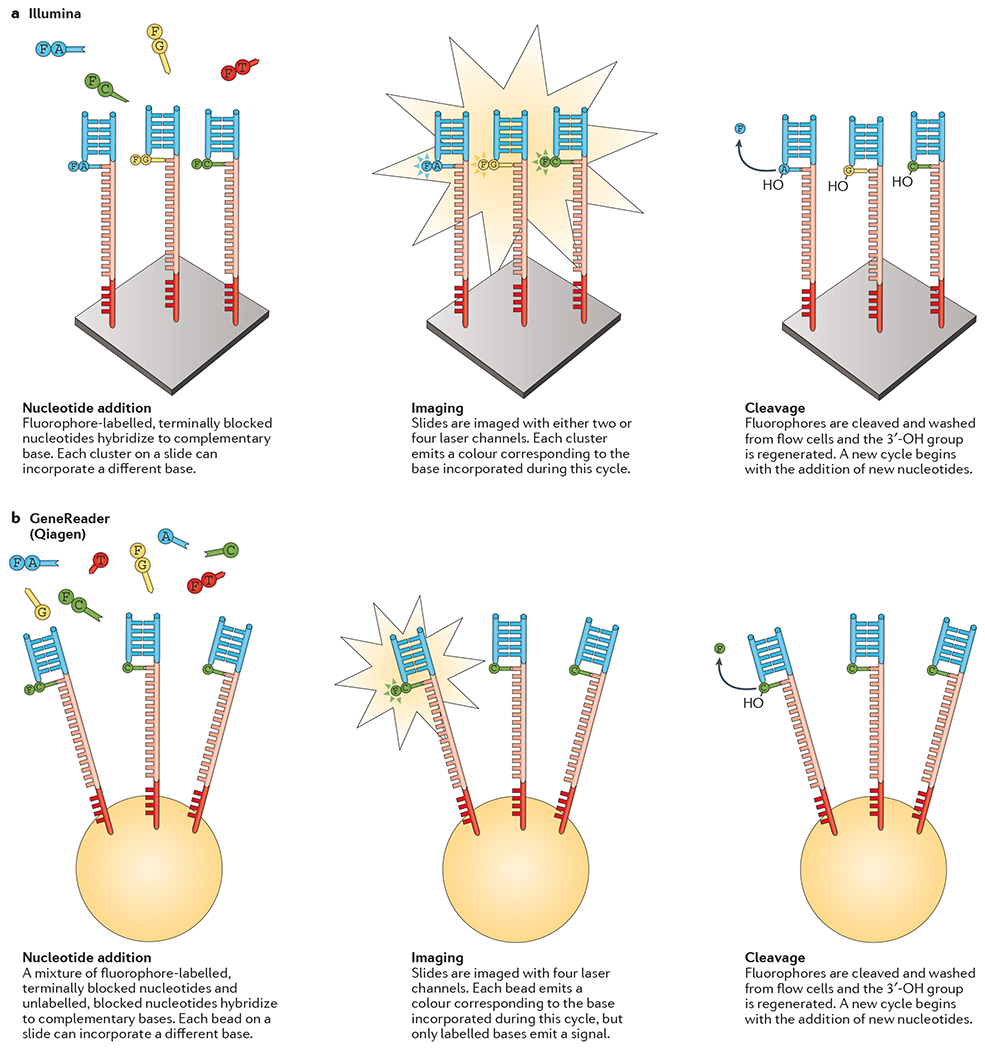
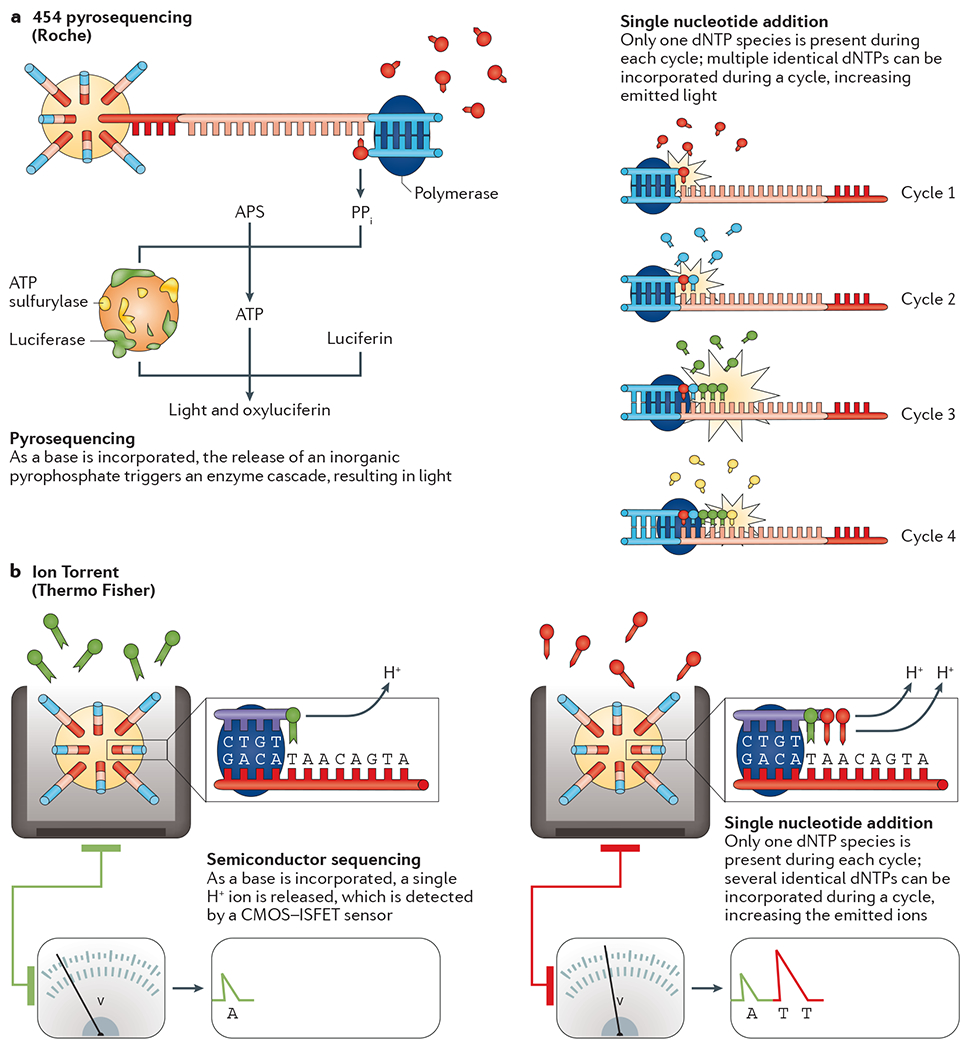
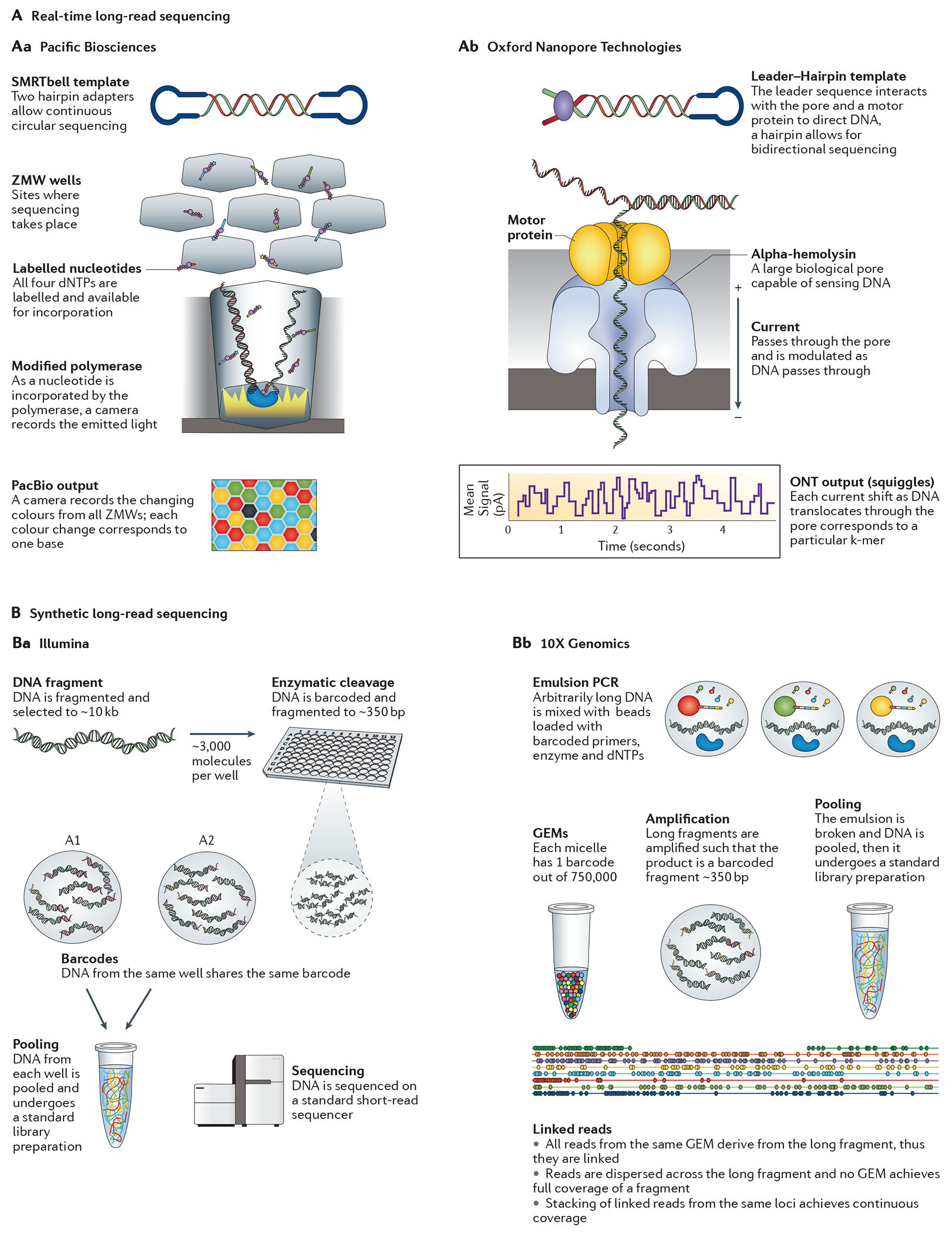
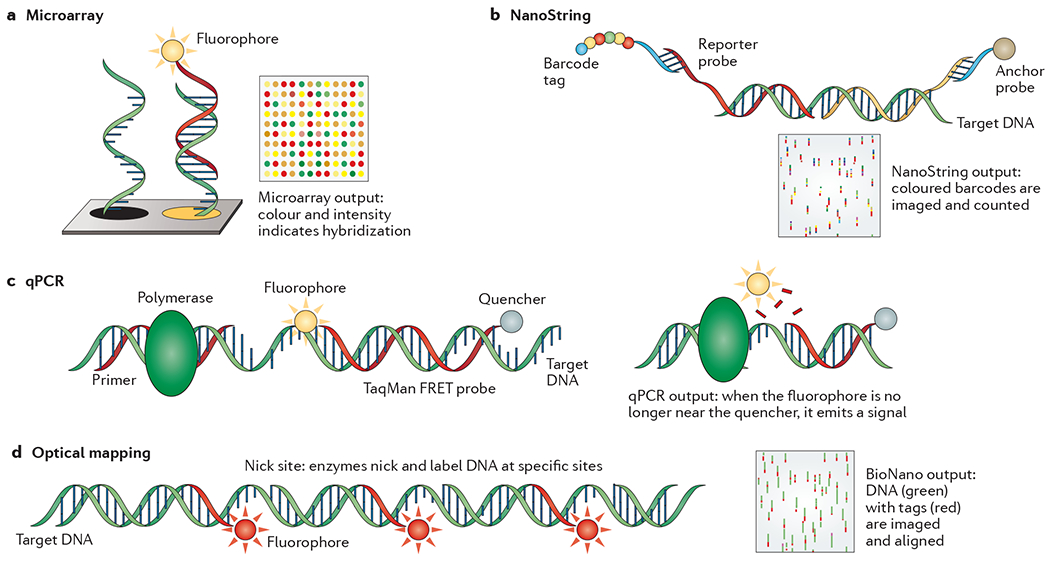
References
-
- Watson JD & Crick FH The structure of DNA. Cold Spring Harb. Symp. Quant. Biol 18, 123–131 (1953). - PubMed
-
- Mardis ER Next-generation sequencing platforms. Annu. Rev. Anal. Chem. (Palo Alto Calif.) 6, 287–303 (2013). - PubMed
-
This article provides a concise description of technological advancements supporting NGS.
-
- Wetterstrand KA DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP). National Human Genome Research Institute; [online], http://www.genome.gov/sequencingcosts (updated 15 Jan 2016).
-
- Kircher M & Kelso J High-throughput DNA sequencing — concepts and limitations. Bioessays 32, 524–536 (2010). - PubMed
-
- Veritas Genetics. Veritas genetics launches $999 whole genome and sets new standard for genetic testing — Press Release. Veritas Genetics; [online], https://www.veritasgenetics.com/documents/VG-launches-999-whole-genome.pdf (updated 4 Mar 2016).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
