

Molecular Characterization of Three Canine Models of Human Rare Bone Diseases: Caffey, van den Ende-Gupta, and Raine Syndromes
- PMID: 27187611
- PMCID: PMC4871343
- DOI: 10.1371/journal.pgen.1006037
Molecular Characterization of Three Canine Models of Human Rare Bone Diseases: Caffey, van den Ende-Gupta, and Raine Syndromes
Abstract
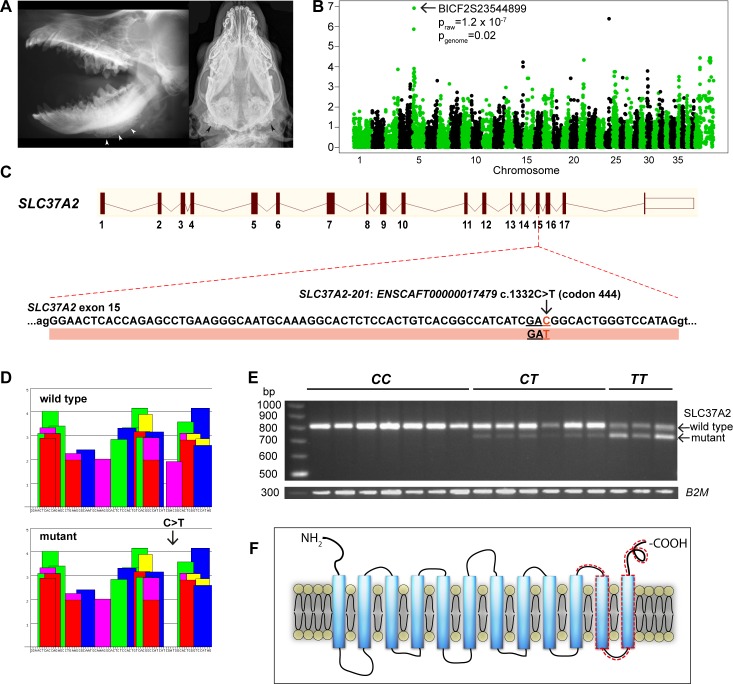
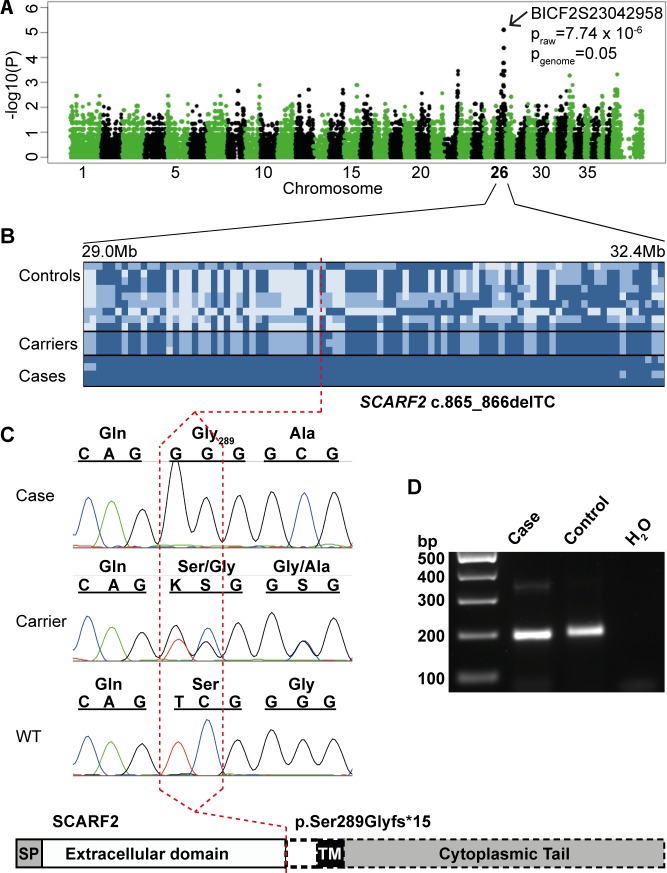
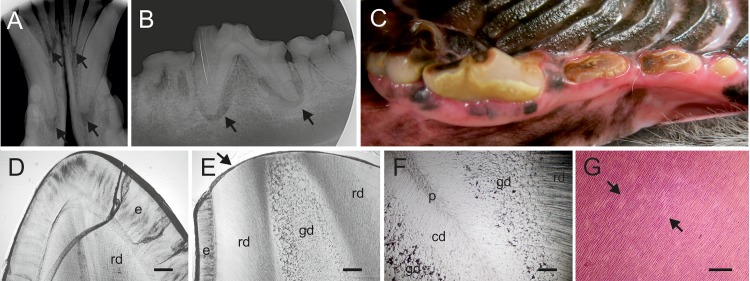
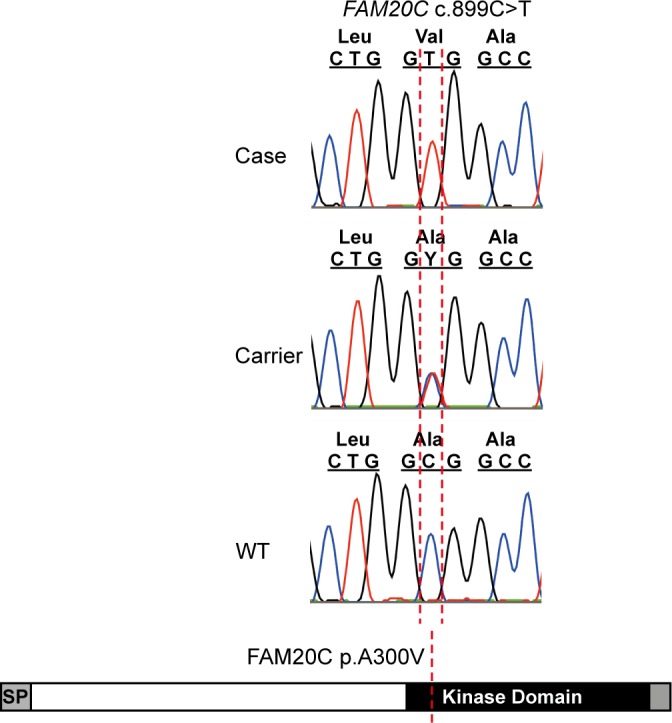
One to two percent of all children are born with a developmental disorder requiring pediatric hospital admissions. For many such syndromes, the molecular pathogenesis remains poorly characterized. Parallel developmental disorders in other species could provide complementary models for human rare diseases by uncovering new candidate genes, improving the understanding of the molecular mechanisms and opening possibilities for therapeutic trials. We performed various experiments, e.g. combined genome-wide association and next generation sequencing, to investigate the clinico-pathological features and genetic causes of three developmental syndromes in dogs, including craniomandibular osteopathy (CMO), a previously undescribed skeletal syndrome, and dental hypomineralization, for which we identified pathogenic variants in the canine SLC37A2 (truncating splicing enhancer variant), SCARF2 (truncating 2-bp deletion) and FAM20C (missense variant) genes, respectively. CMO is a clinical equivalent to an infantile cortical hyperostosis (Caffey disease), for which SLC37A2 is a new candidate gene. SLC37A2 is a poorly characterized member of a glucose-phosphate transporter family without previous disease associations. It is expressed in many tissues, including cells of the macrophage lineage, e.g. osteoclasts, and suggests a disease mechanism, in which an impaired glucose homeostasis in osteoclasts compromises their function in the developing bone, leading to hyperostosis. Mutations in SCARF2 and FAM20C have been associated with the human van den Ende-Gupta and Raine syndromes that include numerous features similar to the affected dogs. Given the growing interest in the molecular characterization and treatment of human rare diseases, our study presents three novel physiologically relevant models for further research and therapy approaches, while providing the molecular identity for the canine conditions.
Conflict of interest statement
I have read the journal's policy and the authors of this manuscript have the following competing interests: Genetic tests will be available from Genoscoper Ltd, which is partly owned by HL. CD is affiliated at the Institute of Genetics, University of Bern, Switzerland, which currently offers a diagnostic genetic test for CMO.
Figures

References
-
- Scriver CR. (1995) Disease, war, and biology: Languages for medicine—and pediatrics. Pediatr Res 38(6): 819–829. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
