

22q11.2 deletion syndrome
- PMID: 27189754
- PMCID: PMC4900471
- DOI: 10.1038/nrdp.2015.71
22q11.2 deletion syndrome
Abstract
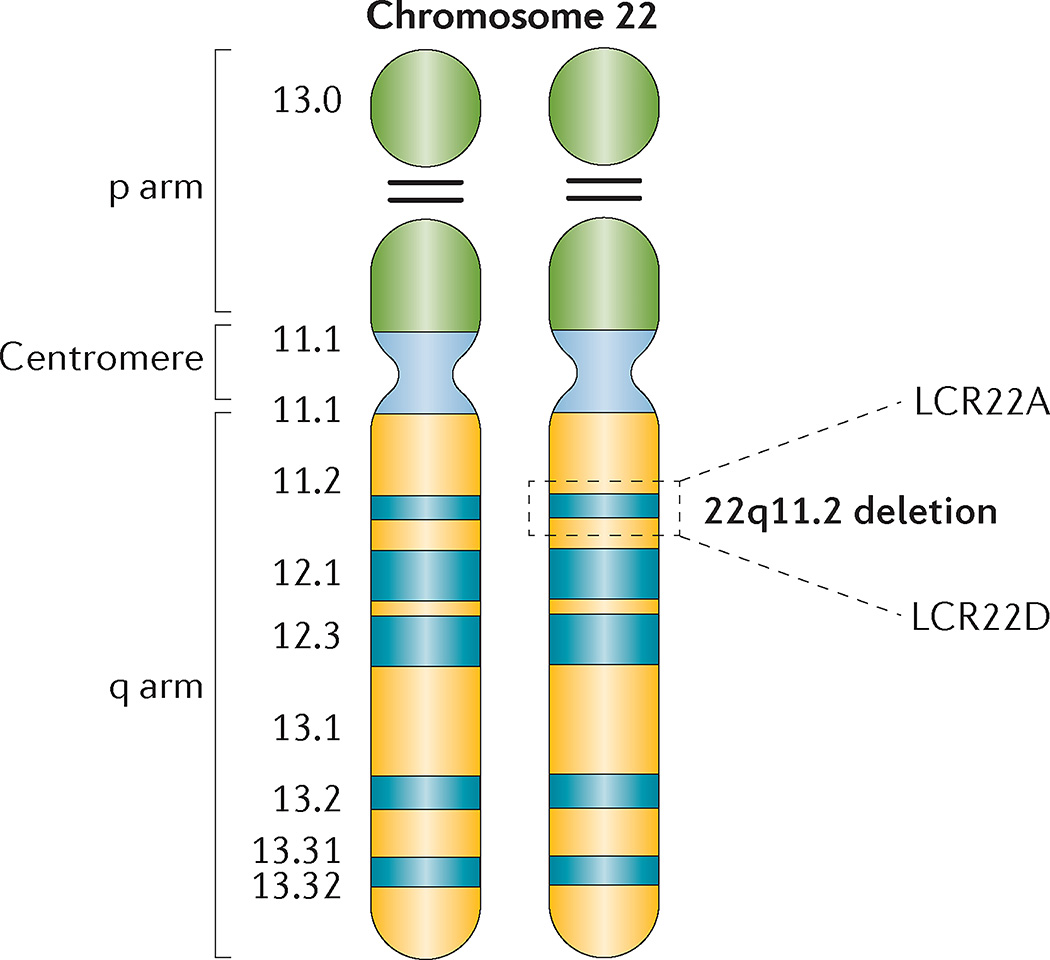
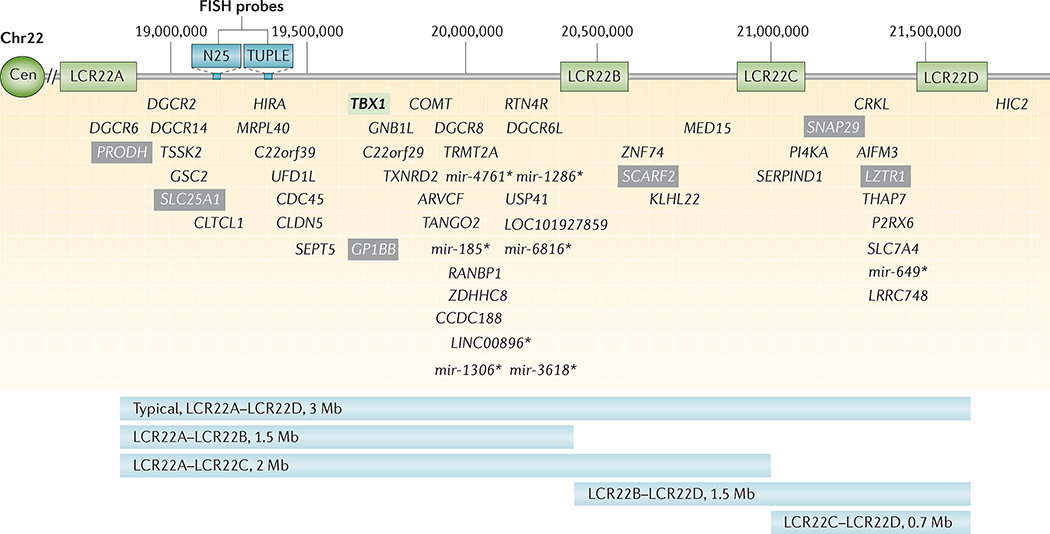
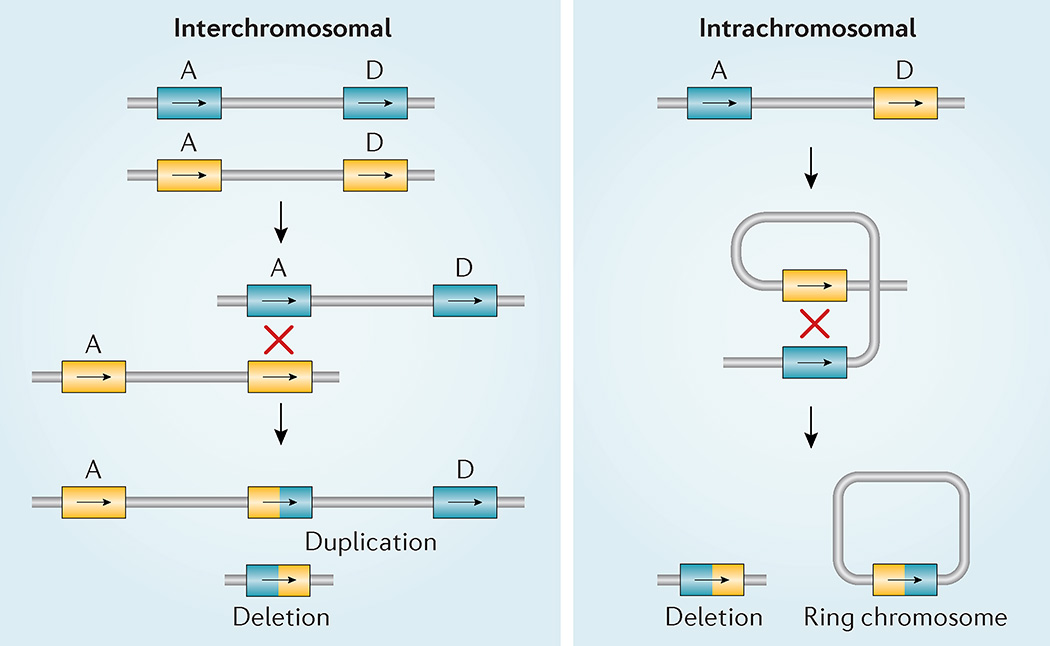
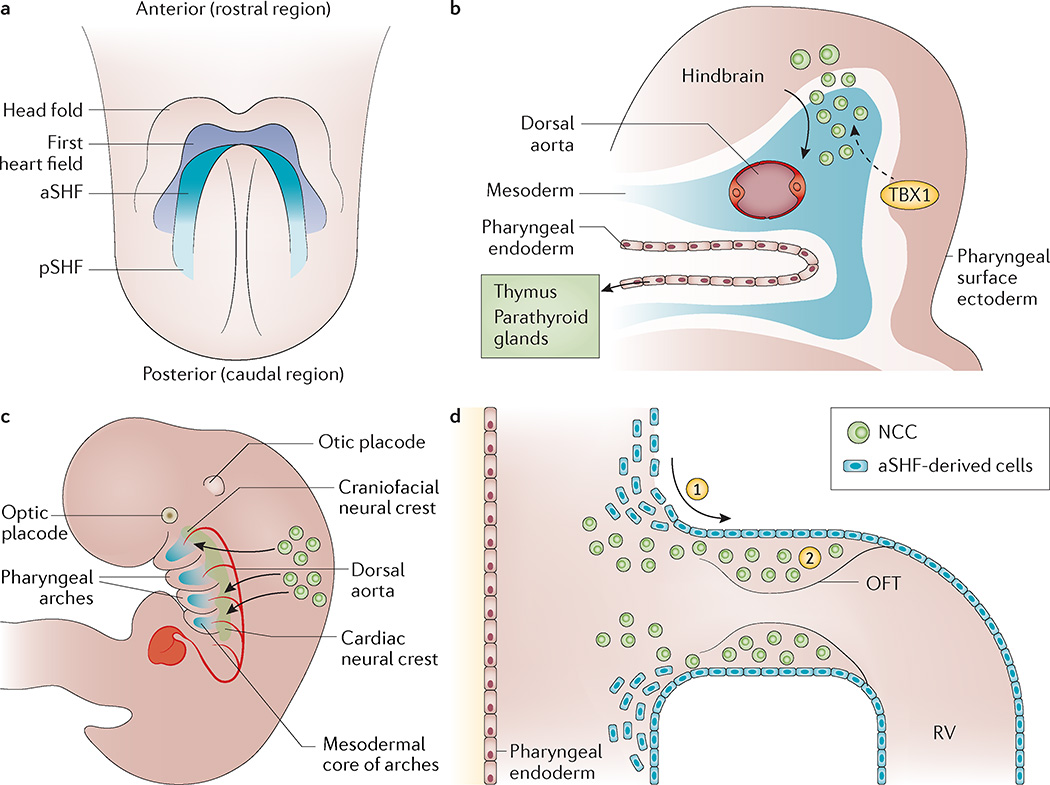
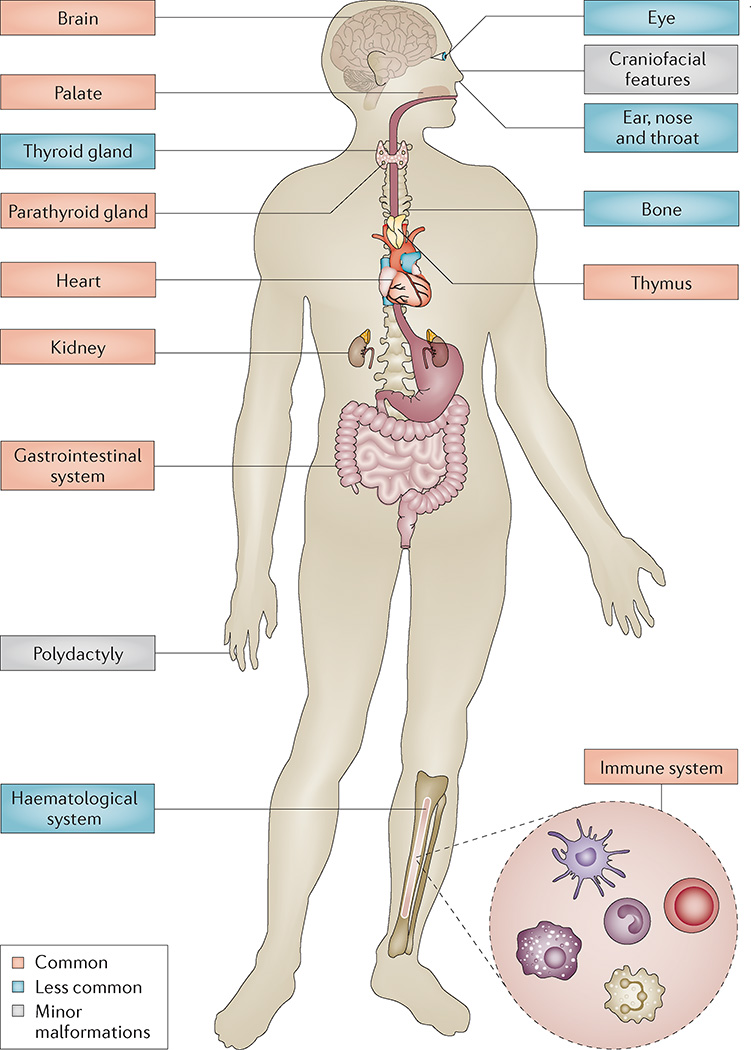
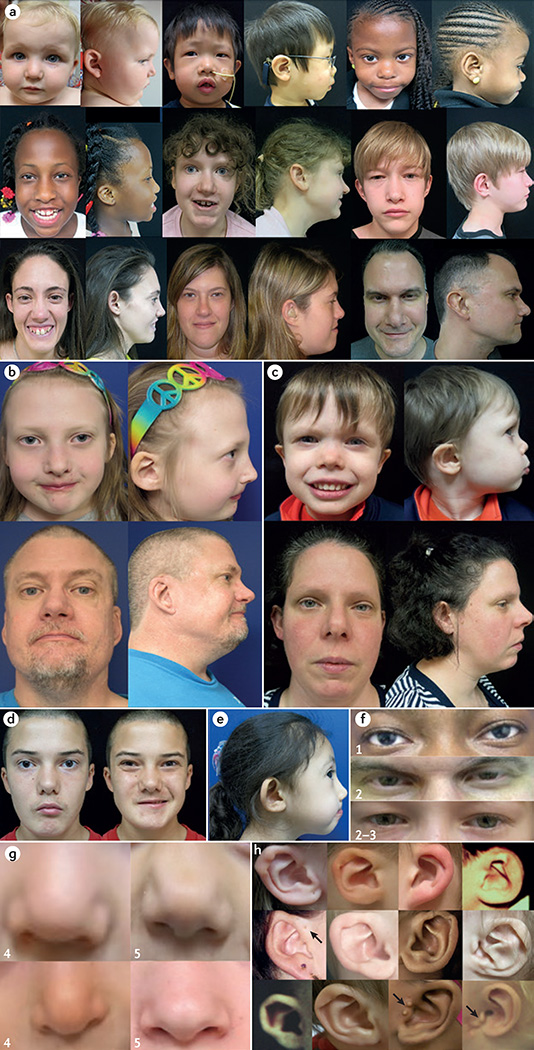
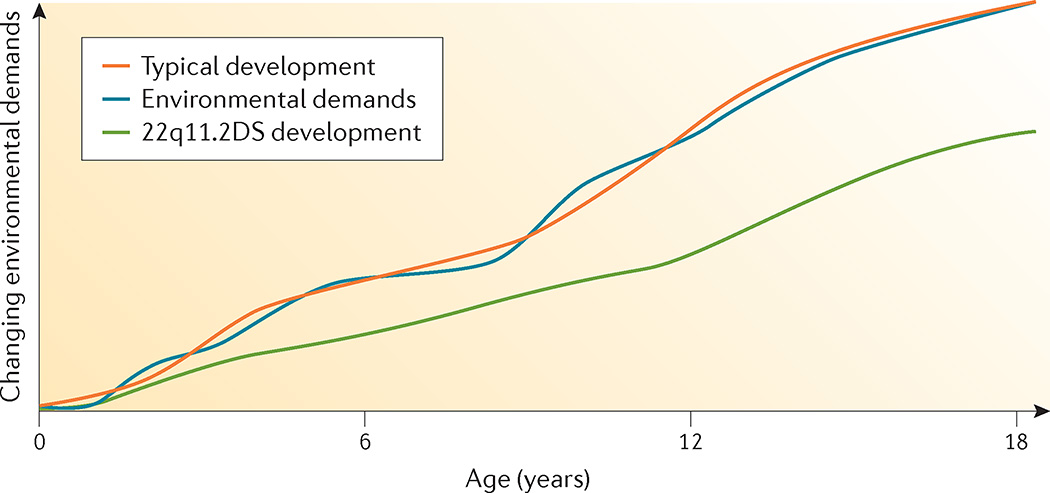
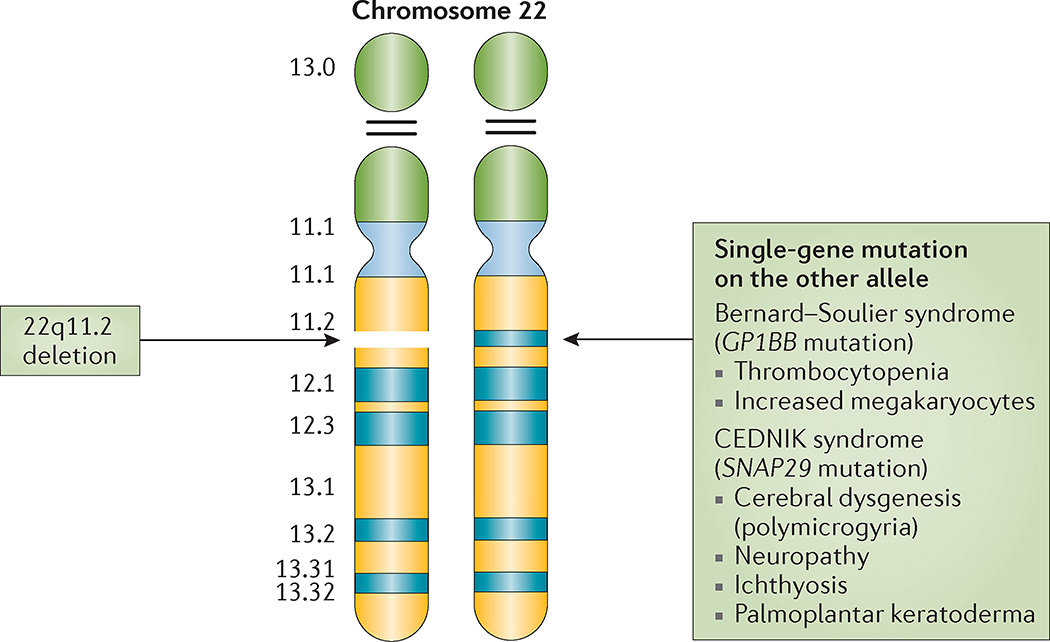
22q11.2 deletion syndrome (22q11.2DS) is the most common chromosomal microdeletion disorder, estimated to result mainly from de novo non-homologous meiotic recombination events occurring in approximately 1 in every 1,000 fetuses. The first description in the English language of the constellation of findings now known to be due to this chromosomal difference was made in the 1960s in children with DiGeorge syndrome, who presented with the clinical triad of immunodeficiency, hypoparathyroidism and congenital heart disease. The syndrome is now known to have a heterogeneous presentation that includes multiple additional congenital anomalies and later-onset conditions, such as palatal, gastrointestinal and renal abnormalities, autoimmune disease, variable cognitive delays, behavioural phenotypes and psychiatric illness - all far extending the original description of DiGeorge syndrome. Management requires a multidisciplinary approach involving paediatrics, general medicine, surgery, psychiatry, psychology, interventional therapies (physical, occupational, speech, language and behavioural) and genetic counselling. Although common, lack of recognition of the condition and/or lack of familiarity with genetic testing methods, together with the wide variability of clinical presentation, delays diagnosis. Early diagnosis, preferably prenatally or neonatally, could improve outcomes, thus stressing the importance of universal screening. Equally important, 22q11.2DS has become a model for understanding rare and frequent congenital anomalies, medical conditions, psychiatric and developmental disorders, and may provide a platform to better understand these disorders while affording opportunities for translational strategies across the lifespan for both patients with 22q11.2DS and those with these associated features in the general population.
Figures
References
-
- DiGeorge A. Discussion on a new concept of the cellular immunology. J. Pediatr. 1965;67:907–908.
-
- Takao A, Ando M, Cho K, Kinouchi A, Murakami Y. In: Etiology and Morphogenesis of Congenital Heart Disease. Van Praagh R, Takao A, editors. Futura Pub. Co.; 1980. pp. 253–269.
-
- Digilio MC, Marino B, Formigari R, Giannotti A. Maternal diabetes causing DiGeorge anomaly and renal agenesis. Am. J. Med. Genet. 1995;55:513–514. - PubMed
-
- Sulik KK, Johnston MC, Daft PA, Russell WE, Dehart DB. Fetal alcohol syndrome and DiGeorge anomaly: critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am. J. Med. Genet. Suppl. 1986;2:97–112. - PubMed
-
- Coberly S, Lammer E, Alashari M. Retinoic acid embryopathy: case report and review of literature. Pediatr. Pathol. Lab. Med. 1996;16:823–836. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
