

Capturing the Phylogeny of Holometabola with Mitochondrial Genome Data and Bayesian Site-Heterogeneous Mixture Models
- PMID: 27189999
- PMCID: PMC4898802
- DOI: 10.1093/gbe/evw086
Capturing the Phylogeny of Holometabola with Mitochondrial Genome Data and Bayesian Site-Heterogeneous Mixture Models
Abstract
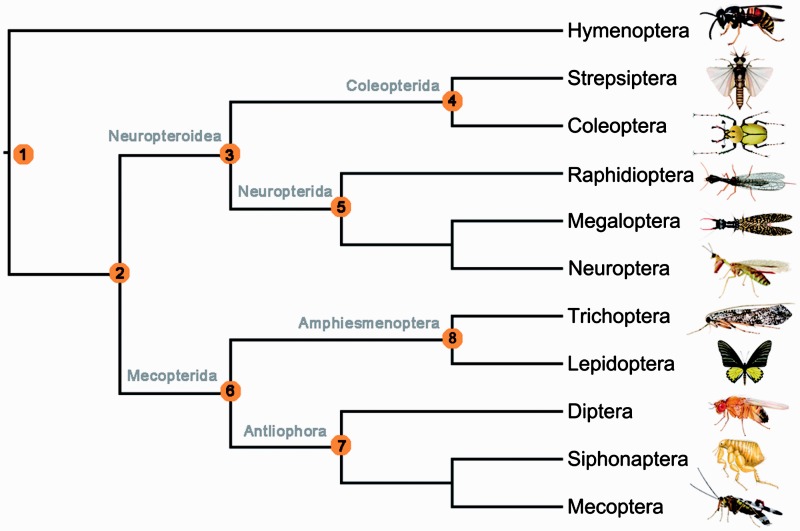
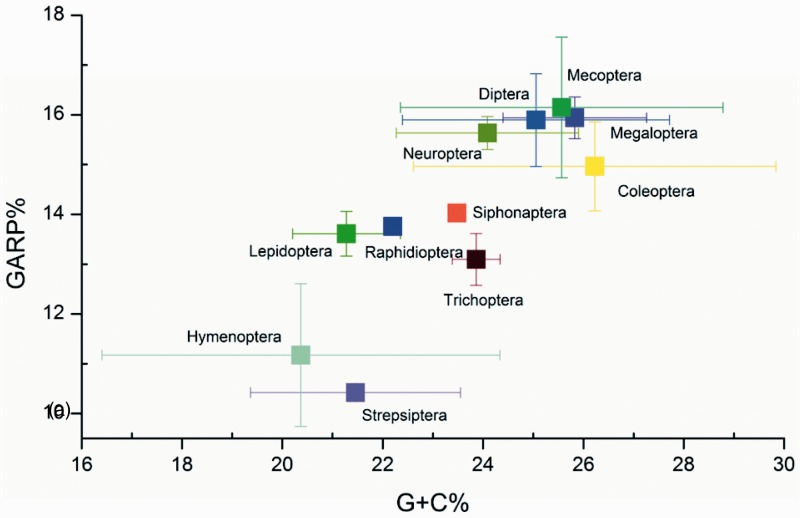
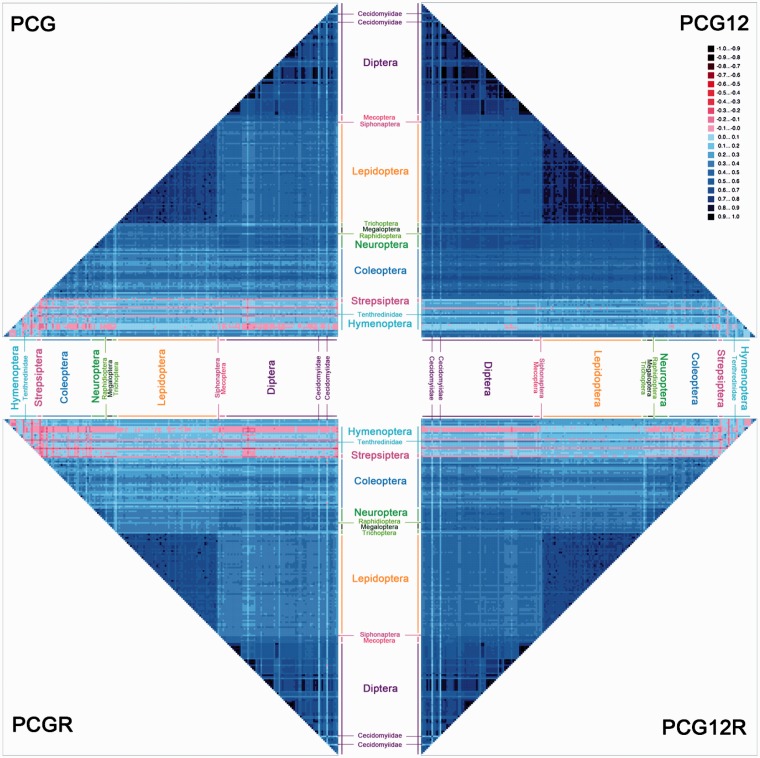
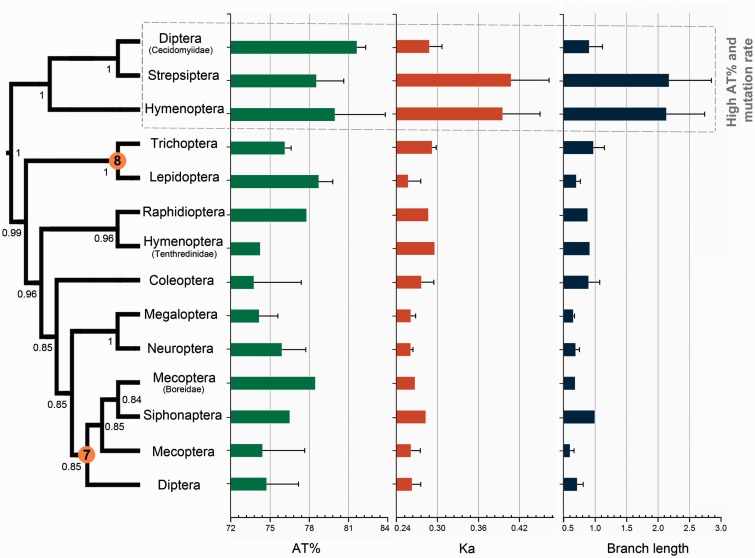
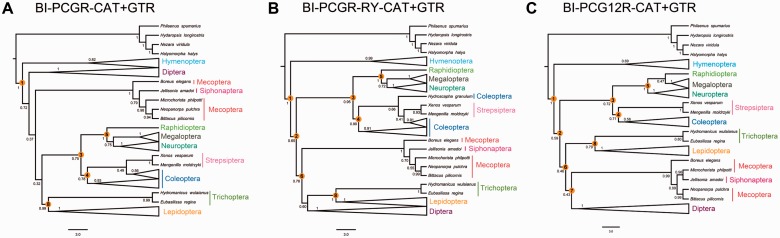
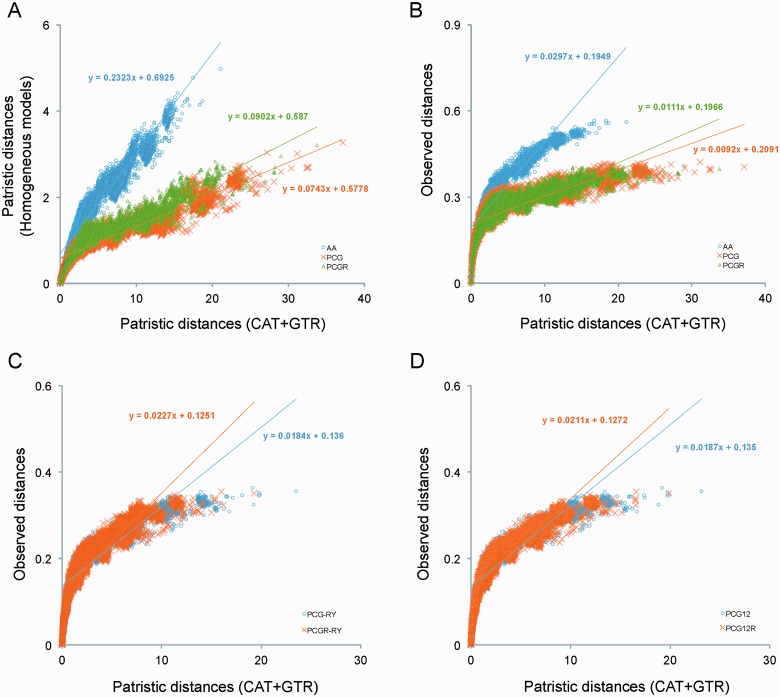
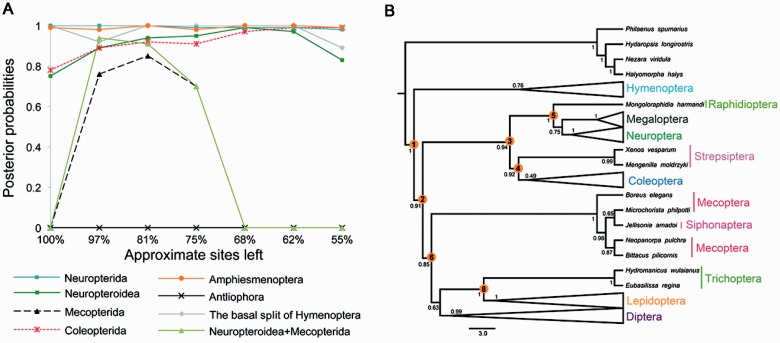
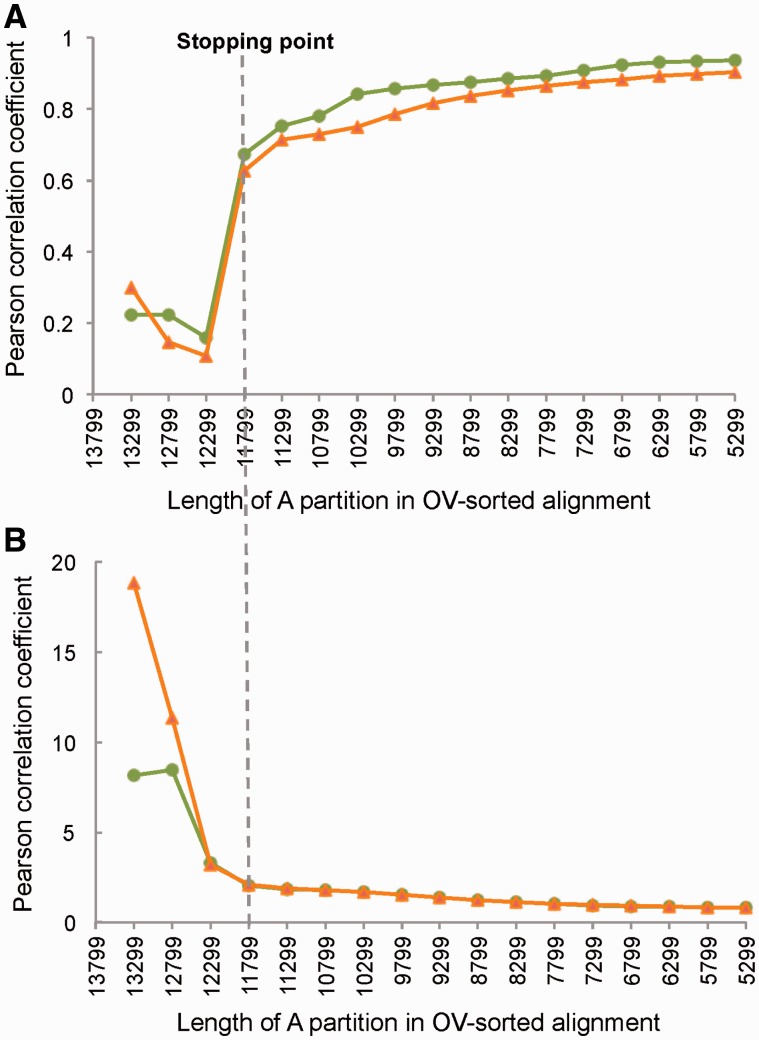
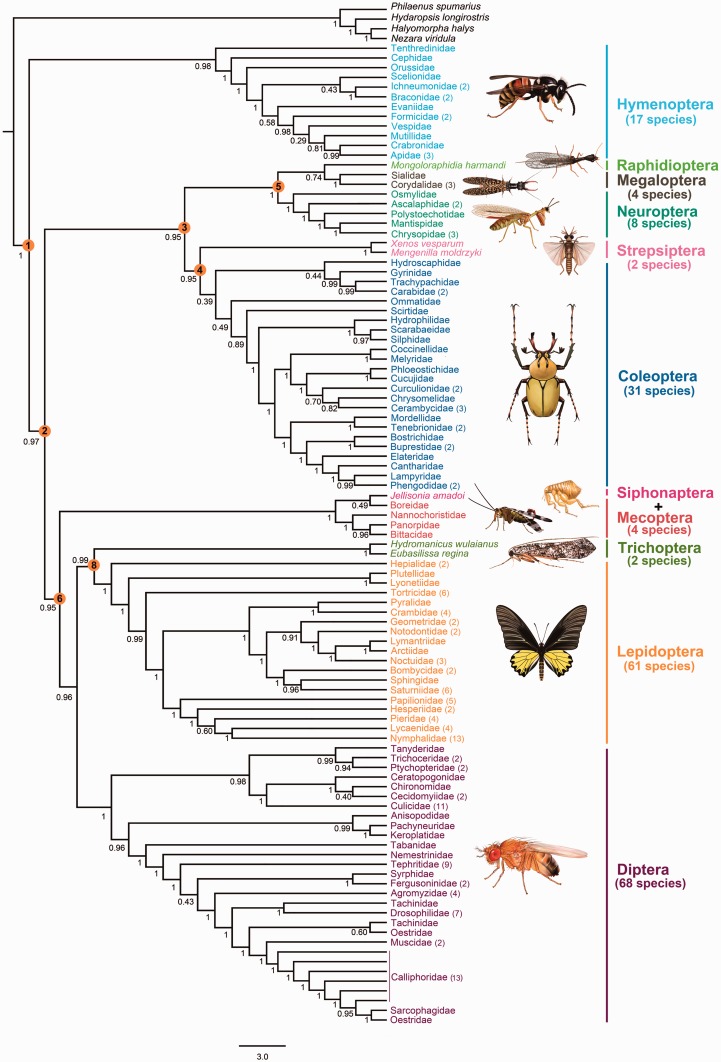
After decades of debate, a mostly satisfactory resolution of relationships among the 11 recognized holometabolan orders of insects has been reached based on nuclear genes, resolving one of the most substantial branches of the tree-of-life, but the relationships are still not well established with mitochondrial genome data. The main reasons have been the absence of sufficient data in several orders and lack of appropriate phylogenetic methods that avoid the systematic errors from compositional and mutational biases in insect mitochondrial genomes. In this study, we assembled the richest taxon sampling of Holometabola to date (199 species in 11 orders), and analyzed both nucleotide and amino acid data sets using several methods. We find the standard Bayesian inference and maximum-likelihood analyses were strongly affected by systematic biases, but the site-heterogeneous mixture model implemented in PhyloBayes avoided the false grouping of unrelated taxa exhibiting similar base composition and accelerated evolutionary rate. The inclusion of rRNA genes and removal of fast-evolving sites with the observed variability sorting method for identifying sites deviating from the mean rates improved the phylogenetic inferences under a site-heterogeneous model, correctly recovering most deep branches of the Holometabola phylogeny. We suggest that the use of mitochondrial genome data for resolving deep phylogenetic relationships requires an assessment of the potential impact of substitutional saturation and compositional biases through data deletion strategies and by using site-heterogeneous mixture models. Our study suggests a practical approach for how to use densely sampled mitochondrial genome data in phylogenetic analyses.
Keywords: Holometabola phylogeny; PhyloBayes; compositional bias; mitochondrial phylogenomics; rate variation; tree-of-life.
© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Abascal F, Posada D, Zardoya R. 2007. MtArt: a new model of amino acid replacement for Arthropoda. Mol Biol Evol. 24:1–5. - PubMed
-
- Baurain D, Brinkmann H, Philippe H. 2007. Lack of resolution in the animal phylogeny: closely spaced cladogeneses or undetected systematic errors? Mol Biol Evol. 24:6–9. - PubMed
-
- Bergsten J. 2005. A review of long-branch attraction. Cladistics 21:163–193. - PubMed
-
- Bernt M, et al. 2013. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol Phylogenet Evol. 69:252–364. - PubMed
MeSH terms
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
