

A platform for the discovery of new macrolide antibiotics
- PMID: 27193679
- PMCID: PMC6526944
- DOI: 10.1038/nature17967
A platform for the discovery of new macrolide antibiotics
Abstract
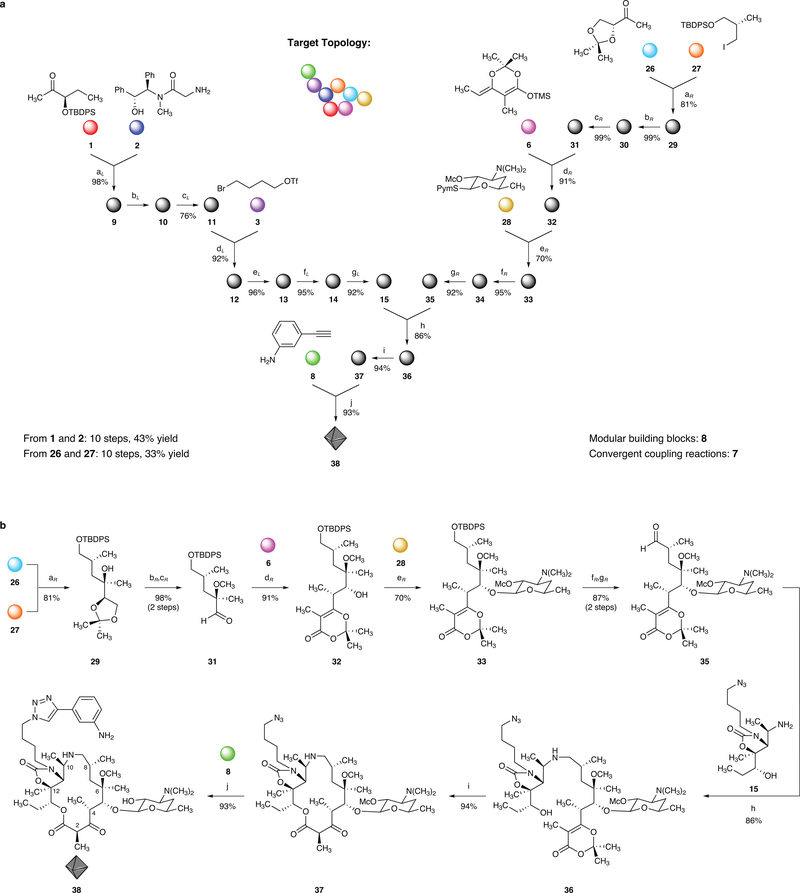
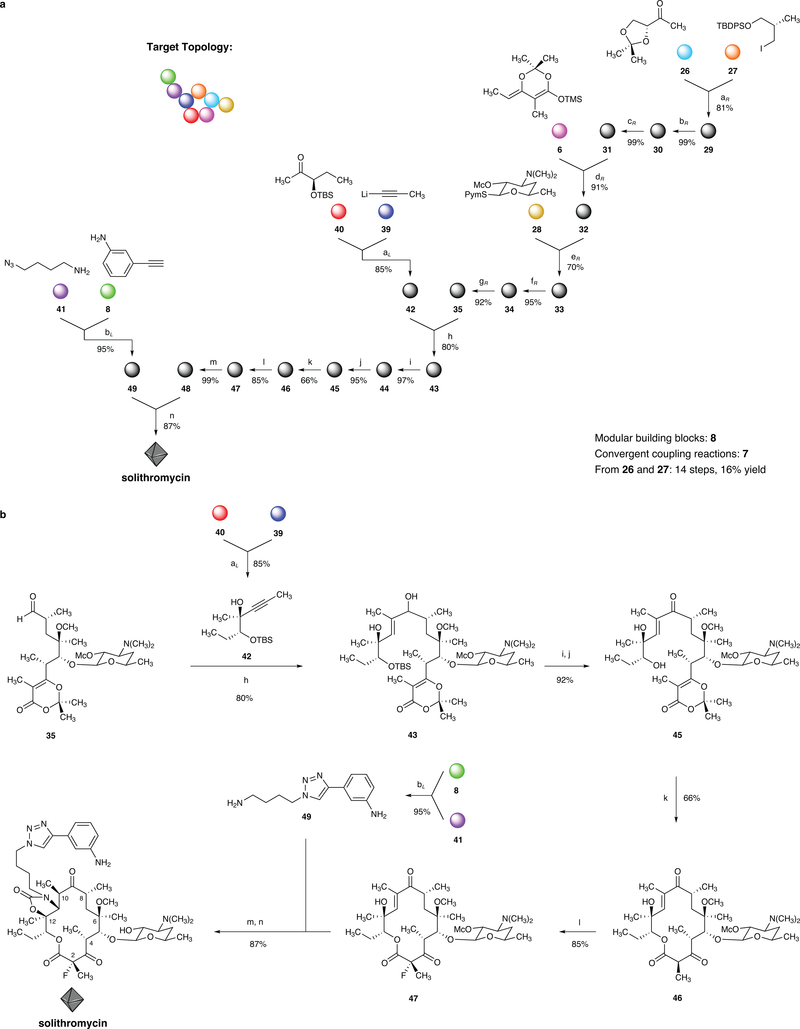
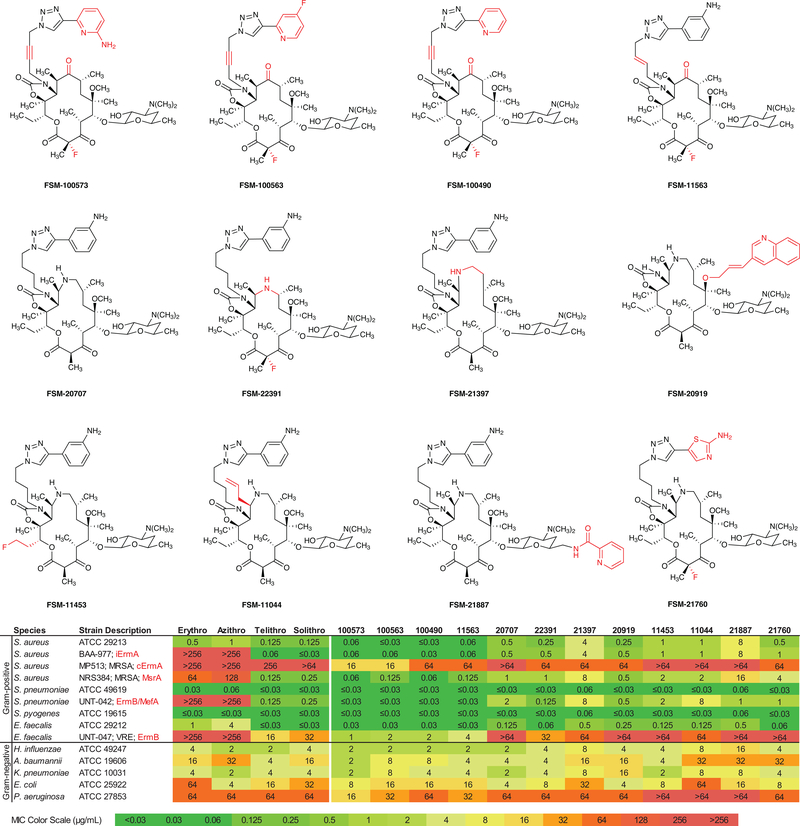
The chemical modification of structurally complex fermentation products, a process known as semisynthesis, has been an important tool in the discovery and manufacture of antibiotics for the treatment of various infectious diseases. However, many of the therapeutics obtained in this way are no longer effective, because bacterial resistance to these compounds has developed. Here we present a practical, fully synthetic route to macrolide antibiotics by the convergent assembly of simple chemical building blocks, enabling the synthesis of diverse structures not accessible by traditional semisynthetic approaches. More than 300 new macrolide antibiotic candidates, as well as the clinical candidate solithromycin, have been synthesized using our convergent approach. Evaluation of these compounds against a panel of pathogenic bacteria revealed that the majority of these structures had antibiotic activity, some efficacious against strains resistant to macrolides in current use. The chemistry we describe here provides a platform for the discovery of new macrolide antibiotics and may also serve as the basis for their manufacture.
Conflict of interest statement
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at
Figures
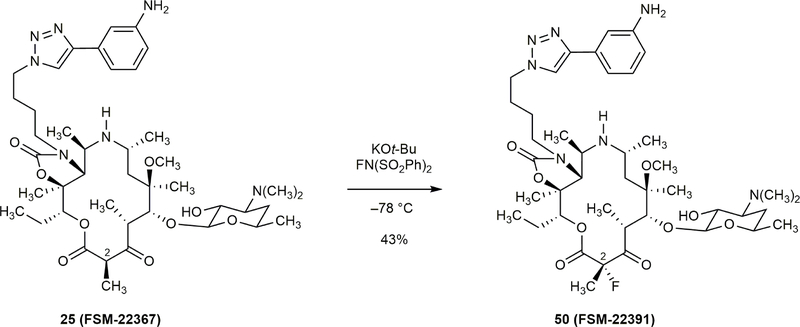
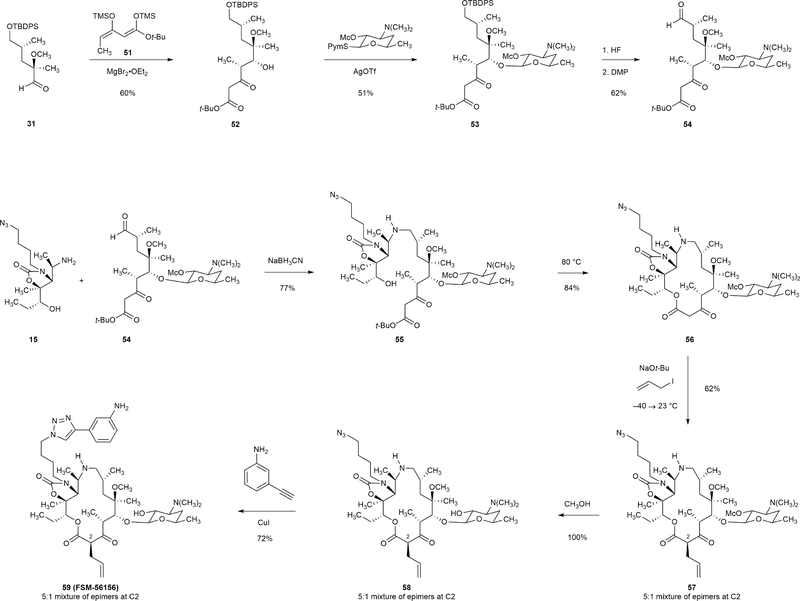
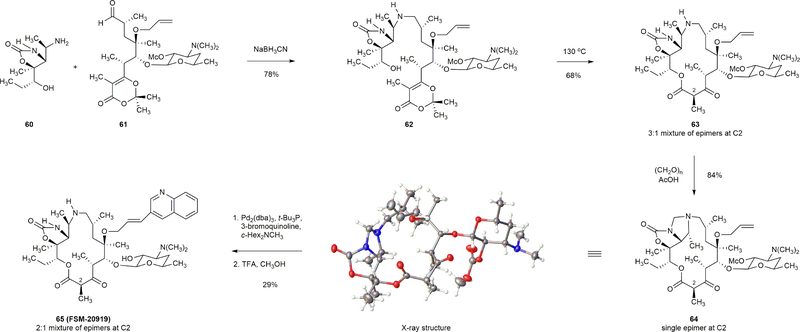
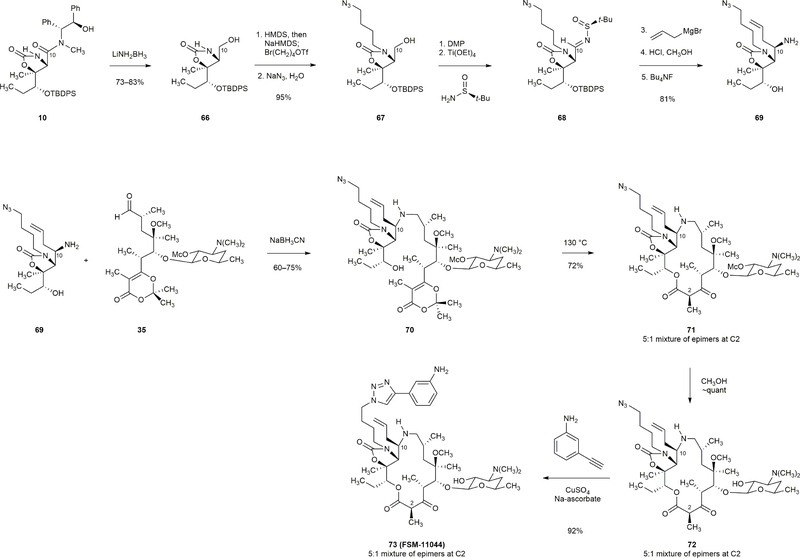
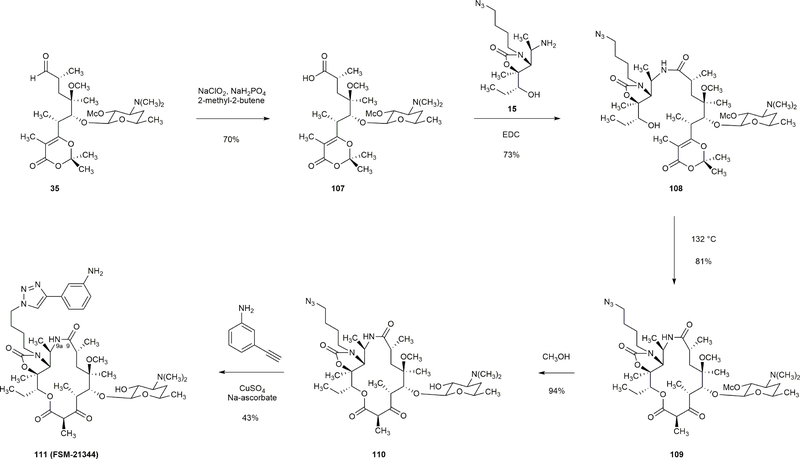
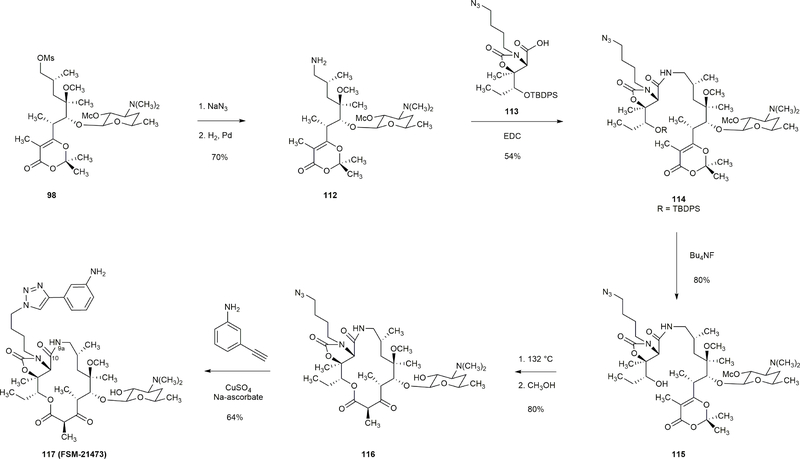
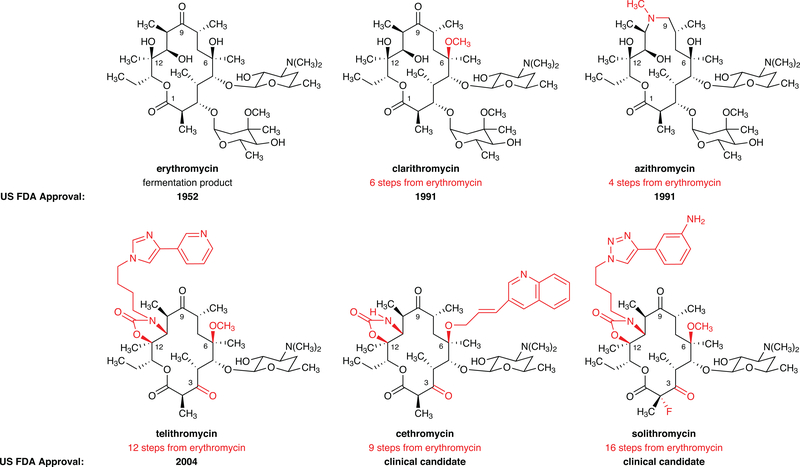
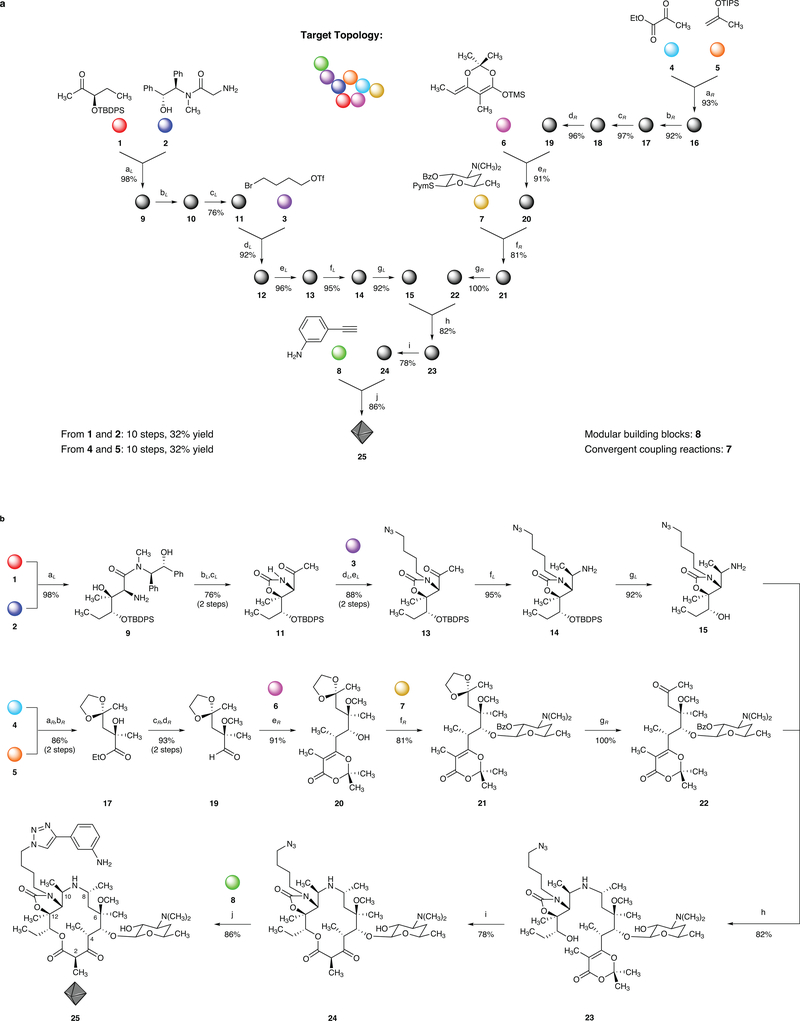
Comment in
-
Drug discovery: Fighting evolution with chemical synthesis.Nature. 2016 May 19;533(7603):326-7. doi: 10.1038/533326a. Nature. 2016. PMID: 27193674 No abstract available.
References
-
- Walsh C Antibiotics: Actions, Origins, Resistance. (American Society for Microbiology Press, 2003).
-
- Mcguire JM et al. Ilotycin, a new antibiotic. Antibiot. Chemother 2, 281–283 (1952). - PubMed
-
- Woodward RB et al. Asymmetric total synthesis of erythromycin. 3. Total synthesis of erythromycin. J. Am. Chem. Soc 103, 3215–3217 (1981).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
