

Primary hyperparathyroidism
- PMID: 27194212
- PMCID: PMC5385896
- DOI: 10.1038/nrdp.2016.33
Primary hyperparathyroidism
Abstract
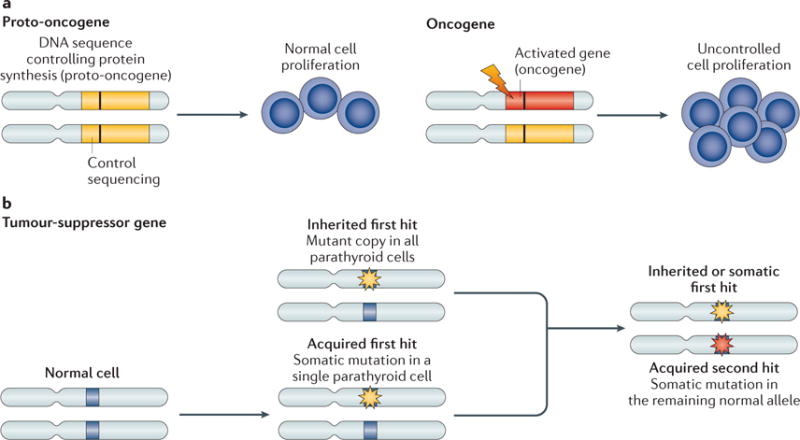
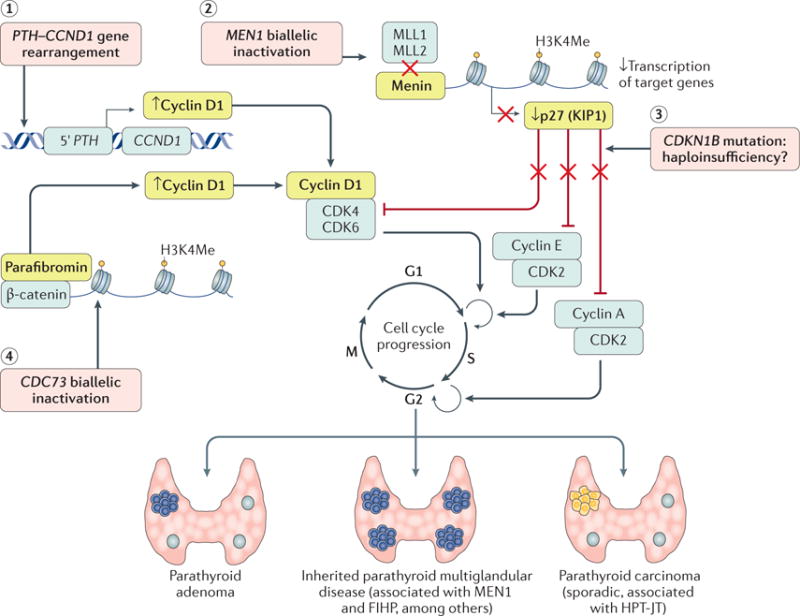
Primary hyperparathyroidism (PHPT) is a common disorder in which parathyroid hormone (PTH) is excessively secreted from one or more of the four parathyroid glands. A single benign parathyroid adenoma is the cause in most people. However, multiglandular disease is not rare and is typically seen in familial PHPT syndromes. The genetics of PHPT is usually monoclonal when a single gland is involved and polyclonal when multiglandular disease is present. The genes that have been implicated in PHPT include proto-oncogenes and tumour-suppressor genes. Hypercalcaemia is the biochemical hallmark of PHPT. Usually, the concentration of PTH is frankly increased but can remain within the normal range, which is abnormal in the setting of hypercalcaemia. Normocalcaemic PHPT, a variant in which the serum calcium level is persistently normal but PTH levels are increased in the absence of an obvious inciting stimulus, is now recognized. The clinical presentation of PHPT varies from asymptomatic disease (seen in countries where biochemical screening is routine) to classic symptomatic disease in which renal and/or skeletal complications are observed. Management guidelines have recently been revised to help the clinician to decide on the merits of a parathyroidectomy or a non-surgical course. This Primer covers these areas with particular attention to the epidemiology, clinical presentations, genetics, evaluation and guidelines for the management of PHPT.
Conflict of interest statement
J.P.B. is a consultant for Merck, Amgen, Shire Pharmaceuticals and Radius, and receives research support from Shire Pharmaceuticals. A.A.K. receives research grants from Amgen and Shire Pharmaceuticals. All other authors declare no competing interests.
Figures
References
-
- Cope O. The study of hyperparathyroidism at the Massachusetts General Hospital. N Engl J Med. 1966;274:1174–1182. - PubMed
-
- Pallan S, Rahman MO, Khan AA. Diagnosis and management of primary hyperparathyroidism. BMJ. 2012;344:e1013. - PubMed
-
- Bilezikian JP, Silverberg SJ. Clinical practice. Asymptomatic primary hyperparathyroidism. N Engl J Med. 2004;350:1746–1751. This paper reports on the changing clinical presentation of PHPT from symptomatic to asymptomatic. - PubMed
-
- Lowe H, McMahon DJ, Rubin MR, Bilezikian JP, Silverberg SJ. Normocalcemic primary hyperparathyroidism: further characterization of a new clinical phenotype. J Clin Endocrinol Metab. 2007;92:3001–3005. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
