

TopoGromacs: Automated Topology Conversion from CHARMM to GROMACS within VMD
- PMID: 27196035
- PMCID: PMC5543333
- DOI: 10.1021/acs.jcim.6b00103
TopoGromacs: Automated Topology Conversion from CHARMM to GROMACS within VMD
Abstract
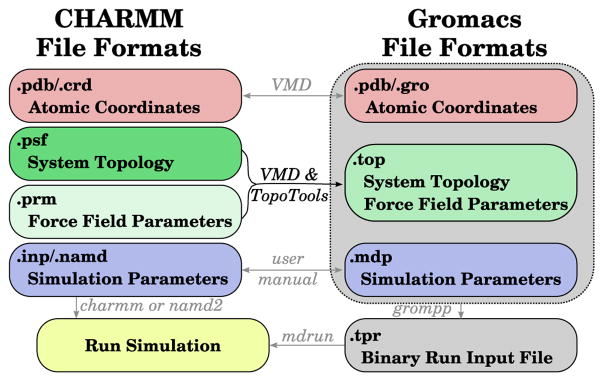
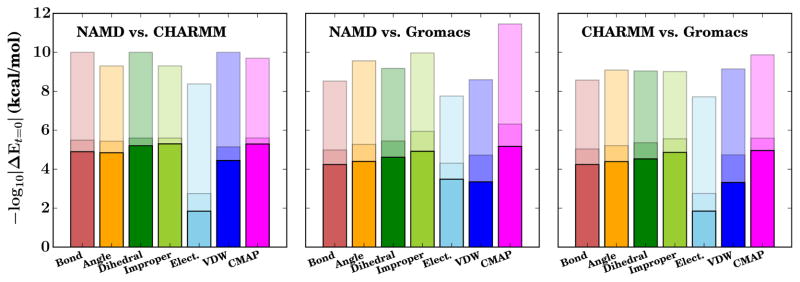
Molecular dynamics (MD) simulation engines use a variety of different approaches for modeling molecular systems with force fields that govern their dynamics and describe their topology. These different approaches introduce incompatibilities between engines, and previously published software bridges the gaps between many popular MD packages, such as between CHARMM and AMBER or GROMACS and LAMMPS. While there are many structure building tools available that generate topologies and structures in CHARMM format, only recently have mechanisms been developed to convert their results into GROMACS input. We present an approach to convert CHARMM-formatted topology and parameters into a format suitable for simulation with GROMACS by expanding the functionality of TopoTools, a plugin integrated within the widely used molecular visualization and analysis software VMD. The conversion process was diligently tested on a comprehensive set of biological molecules in vacuo. The resulting comparison between energy terms shows that the translation performed was lossless as the energies were unchanged for identical starting configurations. By applying the conversion process to conventional benchmark systems that mimic typical modestly sized MD systems, we explore the effect of the implementation choices made in CHARMM, NAMD, and GROMACS. The newly available automatic conversion capability breaks down barriers between simulation tools and user communities and allows users to easily compare simulation programs and leverage their unique features without the tedium of constructing a topology twice.
Figures
Similar articles
-
CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field.J Chem Theory Comput. 2016 Jan 12;12(1):405-13. doi: 10.1021/acs.jctc.5b00935. Epub 2015 Dec 3. J Chem Theory Comput. 2016. PMID: 26631602 Free PMC article.
-
MDWiZ: a platform for the automated translation of molecular dynamics simulations.J Mol Graph Model. 2014 Mar;48:80-6. doi: 10.1016/j.jmgm.2013.12.006. Epub 2013 Dec 25. J Mol Graph Model. 2014. PMID: 24434017
-
Automatic GROMACS topology generation and comparisons of force fields for solvation free energy calculations.J Phys Chem B. 2015 Jan 22;119(3):810-23. doi: 10.1021/jp505332p. Epub 2014 Nov 7. J Phys Chem B. 2015. PMID: 25343332
-
CHARMM-GUI 10 years for biomolecular modeling and simulation.J Comput Chem. 2017 Jun 5;38(15):1114-1124. doi: 10.1002/jcc.24660. Epub 2016 Nov 14. J Comput Chem. 2017. PMID: 27862047 Free PMC article. Review.
-
Comparison of protein force fields for molecular dynamics simulations.Methods Mol Biol. 2008;443:63-88. doi: 10.1007/978-1-59745-177-2_4. Methods Mol Biol. 2008. PMID: 18446282 Review.
Cited by
-
Electrostatic lock in the transport cycle of the multidrug resistance transporter EmrE.Proc Natl Acad Sci U S A. 2018 Aug 7;115(32):E7502-E7511. doi: 10.1073/pnas.1722399115. Epub 2018 Jul 19. Proc Natl Acad Sci U S A. 2018. PMID: 30026196 Free PMC article.
-
Replica-Exchange Enveloping Distribution Sampling Using Generalized AMBER Force-Field Topologies: Application to Relative Hydration Free-Energy Calculations for Large Sets of Molecules.J Chem Inf Model. 2022 Jun 27;62(12):3043-3056. doi: 10.1021/acs.jcim.2c00383. Epub 2022 Jun 8. J Chem Inf Model. 2022. PMID: 35675713 Free PMC article.
-
Assigning crystallographic electron densities with free energy calculations-The case of the fluoride channel Fluc.PLoS One. 2018 May 17;13(5):e0196751. doi: 10.1371/journal.pone.0196751. eCollection 2018. PLoS One. 2018. PMID: 29771936 Free PMC article.
-
Passive membrane transport of lignin-related compounds.Proc Natl Acad Sci U S A. 2019 Nov 12;116(46):23117-23123. doi: 10.1073/pnas.1904643116. Epub 2019 Oct 28. Proc Natl Acad Sci U S A. 2019. PMID: 31659054 Free PMC article.
-
Understanding Lignin Dissolution with Urea and the Formation of a Lignin Nano-Aggregate: A Multiscale Approach.Nanomaterials (Basel). 2024 Mar 27;14(7):593. doi: 10.3390/nano14070593. Nanomaterials (Basel). 2024. PMID: 38607127 Free PMC article.
References
-
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009;30:1545–1614. - PMC - PubMed
-
- Crowley MF, Williamson MJ, Walker RC. CHAMBER: Comprehensive Support for CHARMM Force Fields Within the AMBER Software. Int J Quantum Chem. 2009;109:3767–3772.
-
- Rusu VH, Horta VAC, Horta BAC, Lins RD, Baron R. MDWiZ: A Platform for the Automated Translation of Molecular Dynamics Simulations. J Mol Graph Model. 2014;48:80–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
