

Selection and explosive growth alter genetic architecture and hamper the detection of causal rare variants
- PMID: 27197206
- PMCID: PMC4937562
- DOI: 10.1101/gr.202440.115
Selection and explosive growth alter genetic architecture and hamper the detection of causal rare variants
Abstract
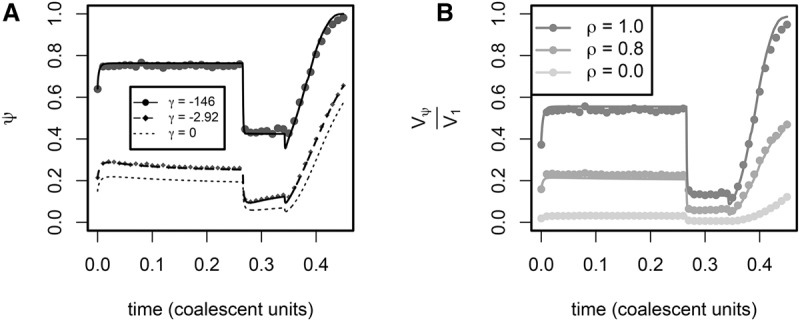
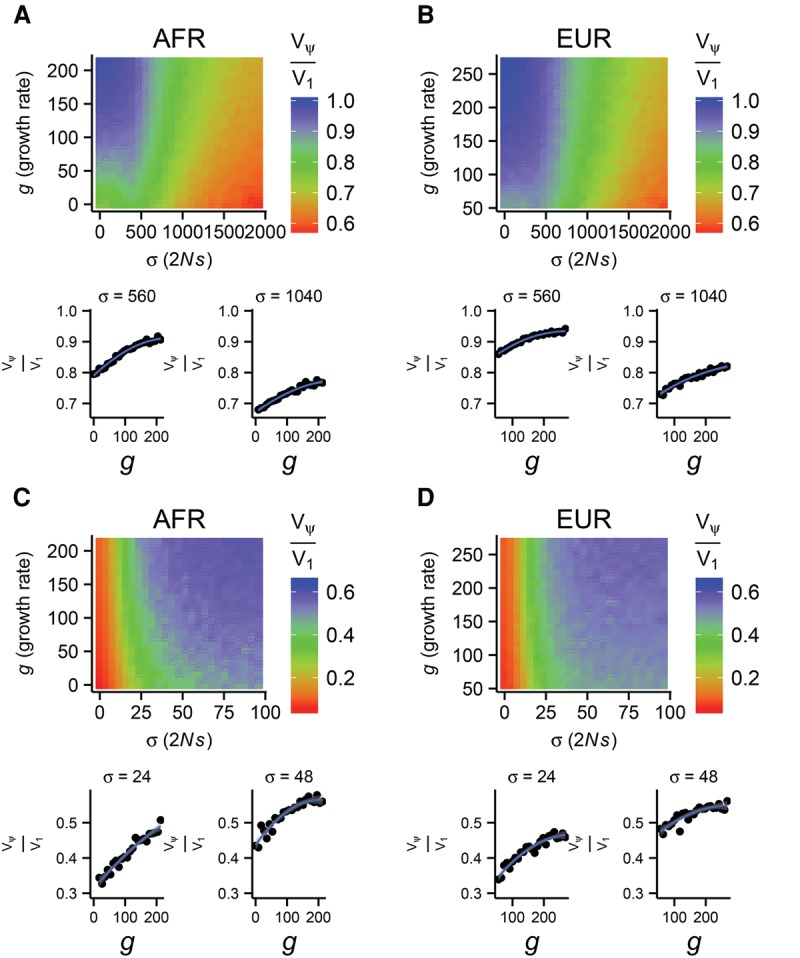
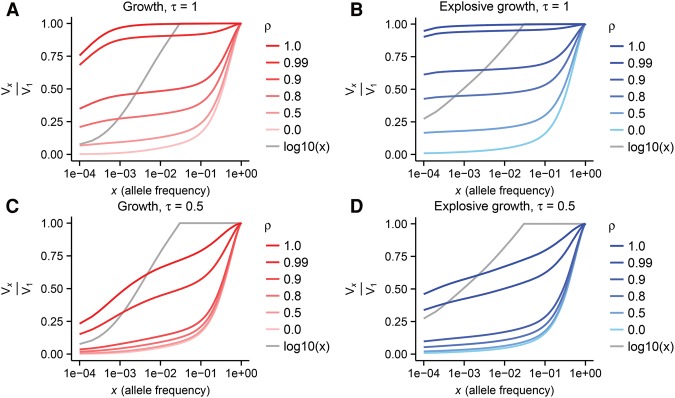
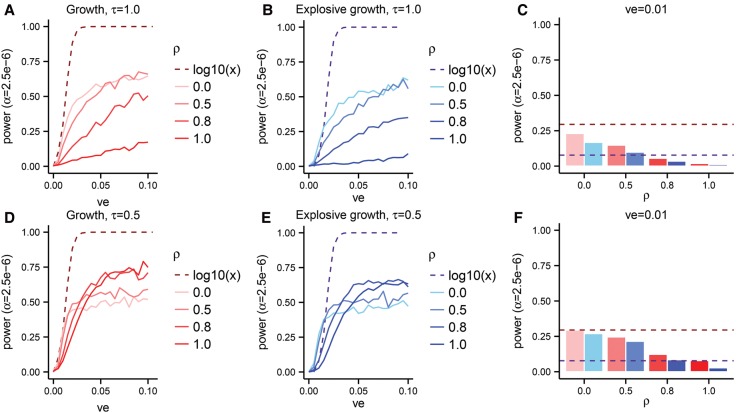
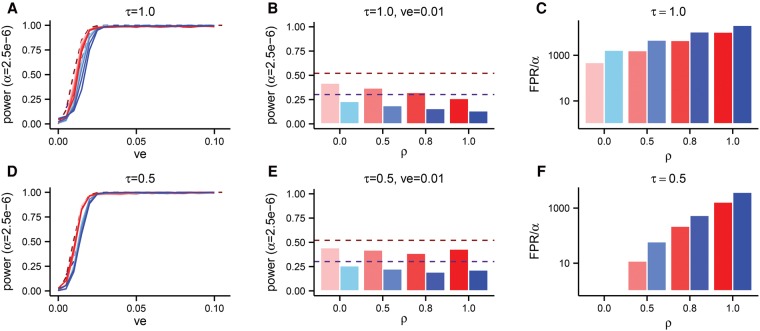
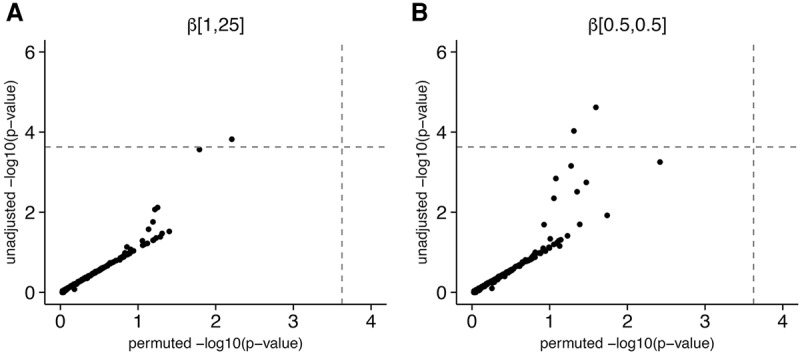
The role of rare alleles in complex phenotypes has been hotly debated, but most rare variant association tests (RVATs) do not account for the evolutionary forces that affect genetic architecture. Here, we use simulation and numerical algorithms to show that explosive population growth, as experienced by human populations, can dramatically increase the impact of very rare alleles on trait variance. We then assess the ability of RVATs to detect causal loci using simulations and human RNA-seq data. Surprisingly, we find that statistical performance is worst for phenotypes in which genetic variance is due mainly to rare alleles, and explosive population growth decreases power. Although many studies have attempted to identify causal rare variants, few have reported novel associations. This has sometimes been interpreted to mean that rare variants make negligible contributions to complex trait heritability. Our work shows that RVATs are not robust to realistic human evolutionary forces, so general conclusions about the impact of rare variants on complex traits may be premature.
© 2016 Uricchio et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources