

Evolutionary assembly patterns of prokaryotic genomes
- PMID: 27197212
- PMCID: PMC4889971
- DOI: 10.1101/gr.200097.115
Evolutionary assembly patterns of prokaryotic genomes
Abstract
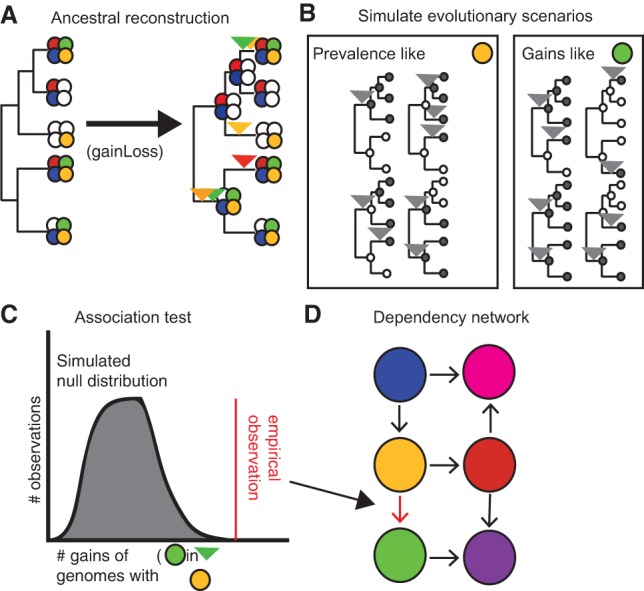
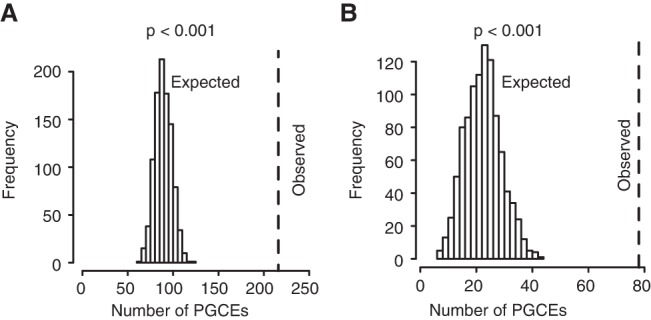
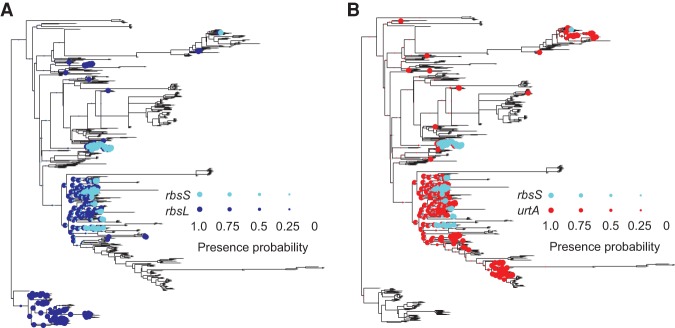
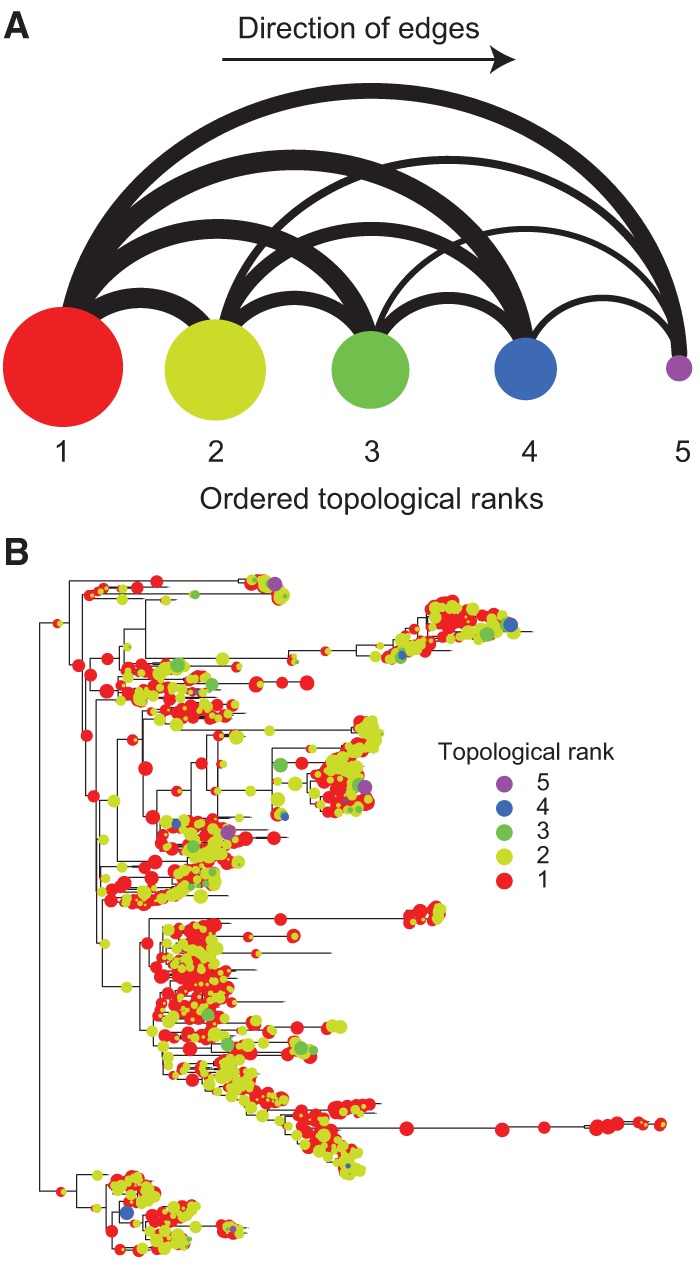
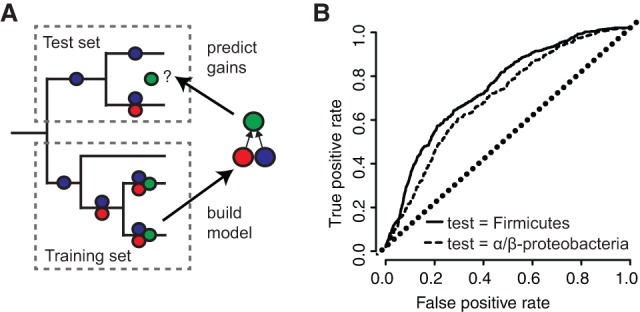
Evolutionary innovation must occur in the context of some genomic background, which limits available evolutionary paths. For example, protein evolution by sequence substitution is constrained by epistasis between residues. In prokaryotes, evolutionary innovation frequently happens by macrogenomic events such as horizontal gene transfer (HGT). Previous work has suggested that HGT can be influenced by ancestral genomic content, yet the extent of such gene-level constraints has not yet been systematically characterized. Here, we evaluated the evolutionary impact of such constraints in prokaryotes, using probabilistic ancestral reconstructions from 634 extant prokaryotic genomes and a novel framework for detecting evolutionary constraints on HGT events. We identified 8228 directional dependencies between genes and demonstrated that many such dependencies reflect known functional relationships, including for example, evolutionary dependencies of the photosynthetic enzyme RuBisCO. Modeling all dependencies as a network, we adapted an approach from graph theory to establish chronological precedence in the acquisition of different genomic functions. Specifically, we demonstrated that specific functions tend to be gained sequentially, suggesting that evolution in prokaryotes is governed by functional assembly patterns. Finally, we showed that these dependencies are universal rather than clade-specific and are often sufficient for predicting whether or not a given ancestral genome will acquire specific genes. Combined, our results indicate that evolutionary innovation via HGT is profoundly constrained by epistasis and historical contingency, similar to the evolution of proteins and phenotypic characters, and suggest that the emergence of specific metabolic and pathological phenotypes in prokaryotes can be predictable from current genomes.
© 2016 Press et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Andam CP, Gogarten JP. 2011. Biased gene transfer in microbial evolution. Nat Rev Microbiol 9: 543–555. - PubMed
-
- Andersson I, Backlund A. 2008. Structure and function of Rubisco. Plant Physiol Biochem 46: 275–291. - PubMed
-
- Baltrus DA. 2013. Exploring the costs of horizontal gene transfer. Trends Ecol Evol 28: 489–495. - PubMed
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources