

SMASH, a fragmentation and sequencing method for genomic copy number analysis
- PMID: 27197213
- PMCID: PMC4889966
- DOI: 10.1101/gr.201491.115
SMASH, a fragmentation and sequencing method for genomic copy number analysis
Abstract
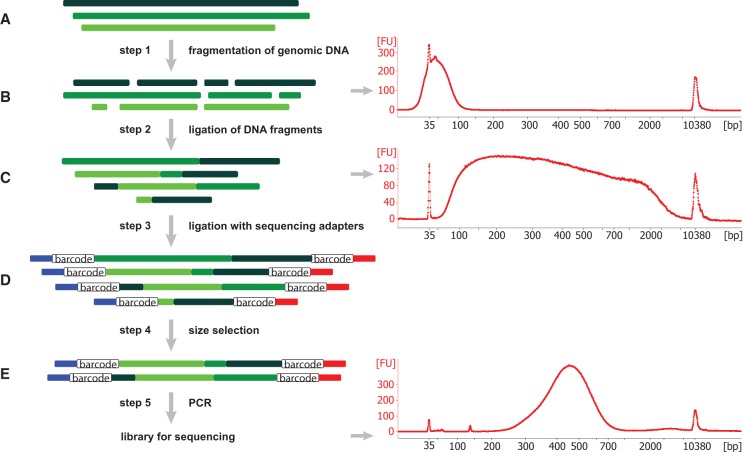
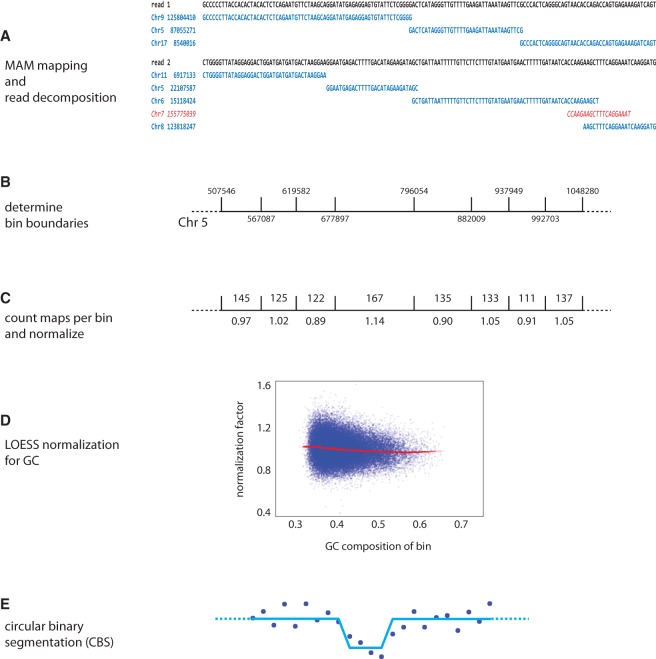
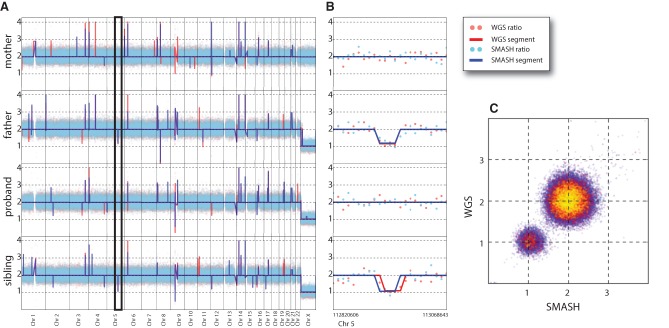
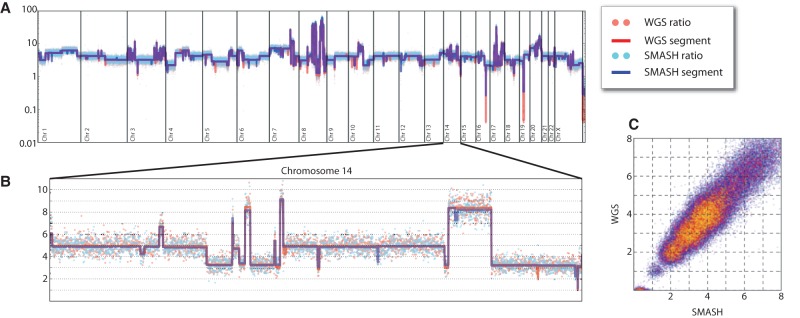
Copy number variants (CNVs) underlie a significant amount of genetic diversity and disease. CNVs can be detected by a number of means, including chromosomal microarray analysis (CMA) and whole-genome sequencing (WGS), but these approaches suffer from either limited resolution (CMA) or are highly expensive for routine screening (both CMA and WGS). As an alternative, we have developed a next-generation sequencing-based method for CNV analysis termed SMASH, for short multiply aggregated sequence homologies. SMASH utilizes random fragmentation of input genomic DNA to create chimeric sequence reads, from which multiple mappable tags can be parsed using maximal almost-unique matches (MAMs). The SMASH tags are then binned and segmented, generating a profile of genomic copy number at the desired resolution. Because fewer reads are necessary relative to WGS to give accurate CNV data, SMASH libraries can be highly multiplexed, allowing large numbers of individuals to be analyzed at low cost. Increased genomic resolution can be achieved by sequencing to higher depth.
© 2016 Wang et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Fischbach GD, Lord C. 2010. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68: 192–195. - PubMed
-
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. 2004. Detection of large-scale variation in the human genome. Nat Genet 36: 949–951. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials