

Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks
- PMID: 27197224
- PMCID: PMC4937568
- DOI: 10.1101/gr.200535.115
Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks
Abstract
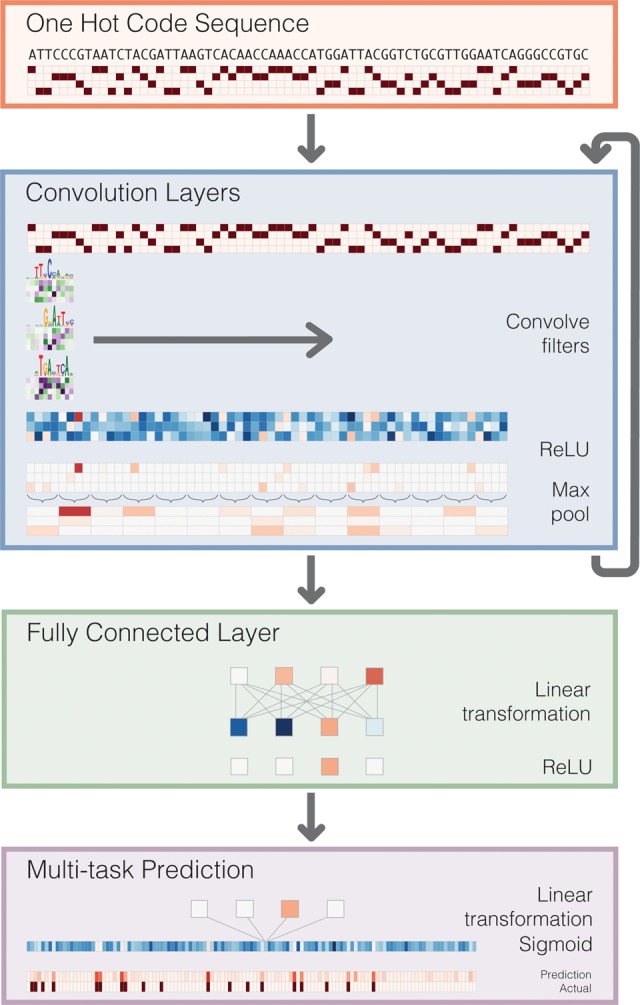
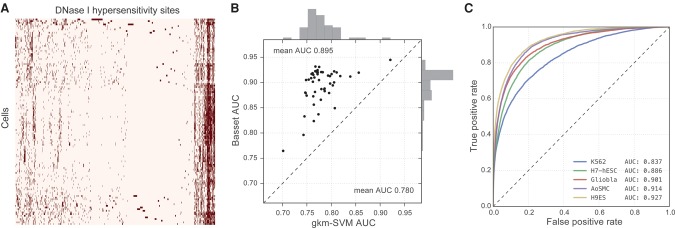
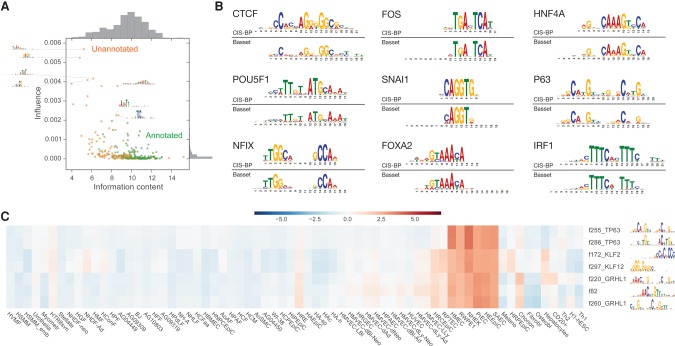
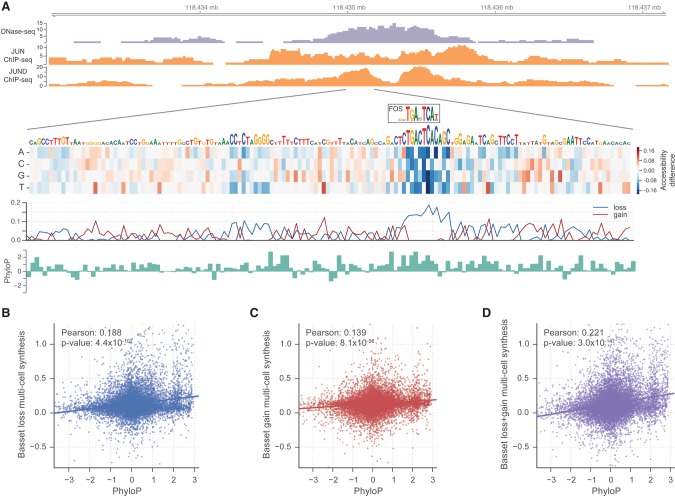
The complex language of eukaryotic gene expression remains incompletely understood. Despite the importance suggested by many noncoding variants statistically associated with human disease, nearly all such variants have unknown mechanisms. Here, we address this challenge using an approach based on a recent machine learning advance-deep convolutional neural networks (CNNs). We introduce the open source package Basset to apply CNNs to learn the functional activity of DNA sequences from genomics data. We trained Basset on a compendium of accessible genomic sites mapped in 164 cell types by DNase-seq, and demonstrate greater predictive accuracy than previous methods. Basset predictions for the change in accessibility between variant alleles were far greater for Genome-wide association study (GWAS) SNPs that are likely to be causal relative to nearby SNPs in linkage disequilibrium with them. With Basset, a researcher can perform a single sequencing assay in their cell type of interest and simultaneously learn that cell's chromatin accessibility code and annotate every mutation in the genome with its influence on present accessibility and latent potential for accessibility. Thus, Basset offers a powerful computational approach to annotate and interpret the noncoding genome.
© 2016 Kelley et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Alipanahi B, Delong A, Weirauch MT, Frey BJ. 2015. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 33: 831–838. - PubMed
-
- Beer MA, Tavazoie S. 2004. Predicting gene expression from sequence. Cell 117: 185–198. - PubMed
-
- Bengio Y, Courville A, Vincent P. 2013. Representation learning: a review and new perspectives. IEEE Trans Pattern Anal Mach Intell 35: 1798–1828. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources