

The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin
- PMID: 27197267
- PMCID: PMC4874568
- DOI: 10.1158/0008-5472.CAN-16-0186
The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin
Abstract
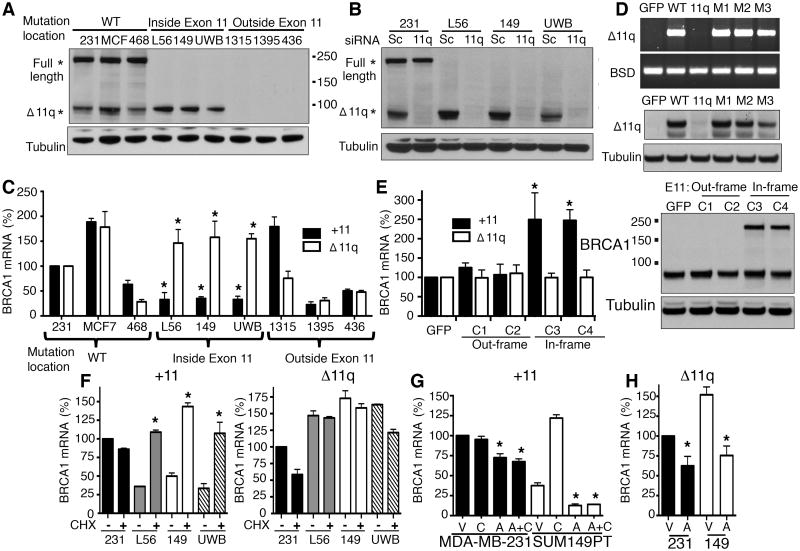
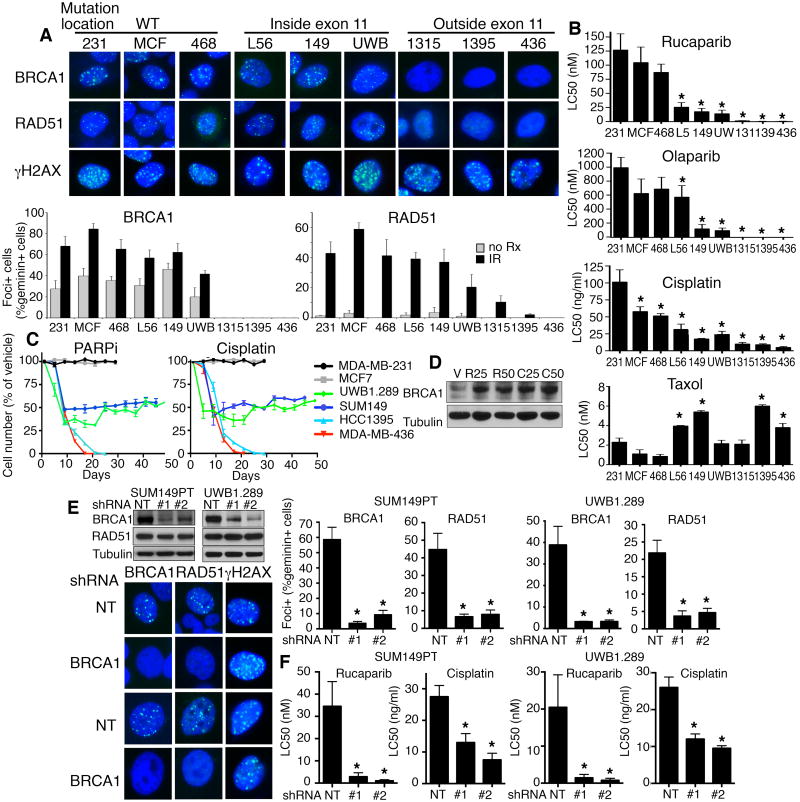
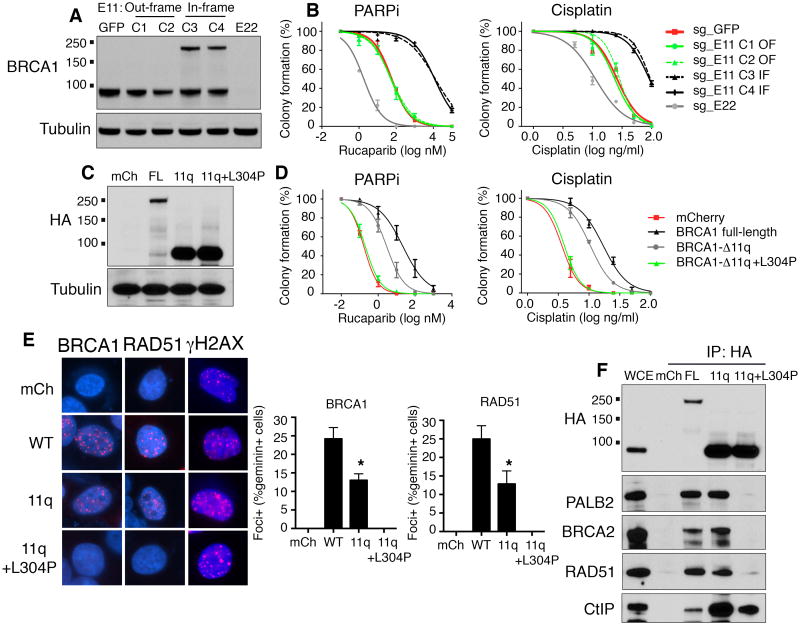
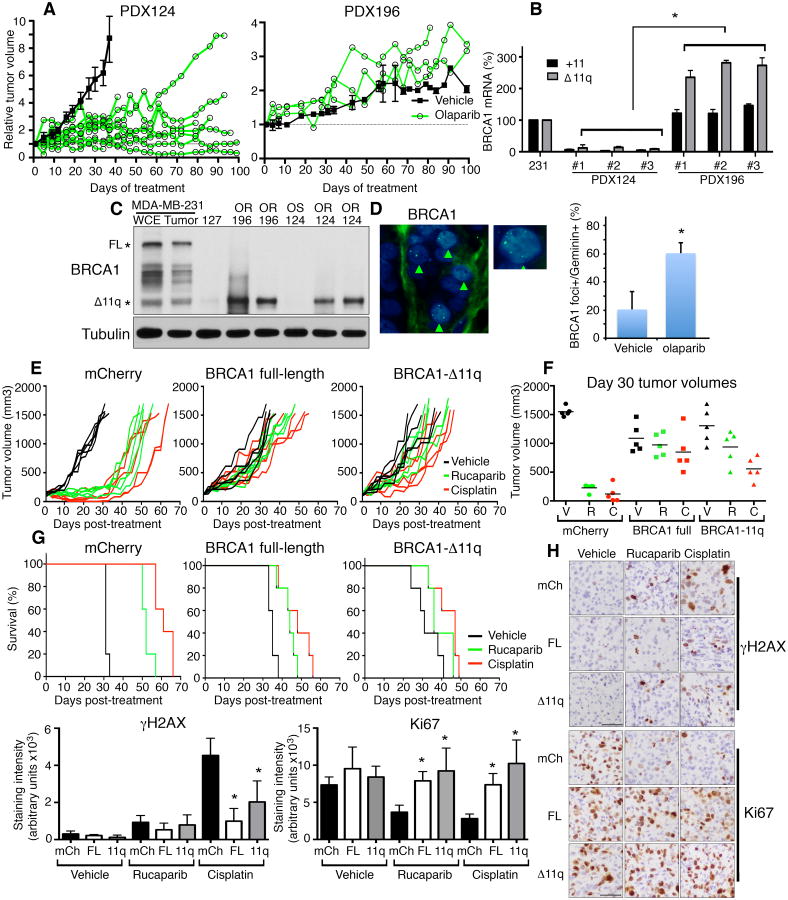
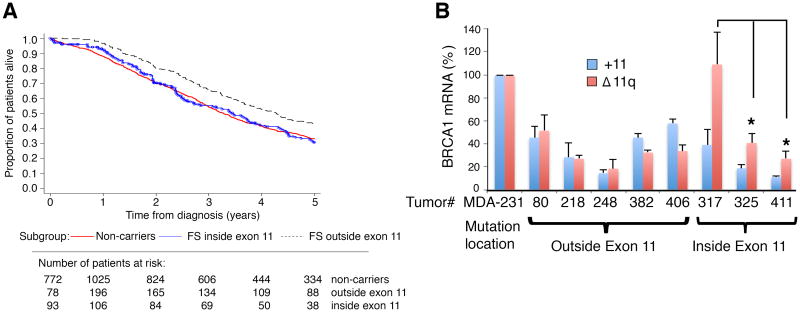
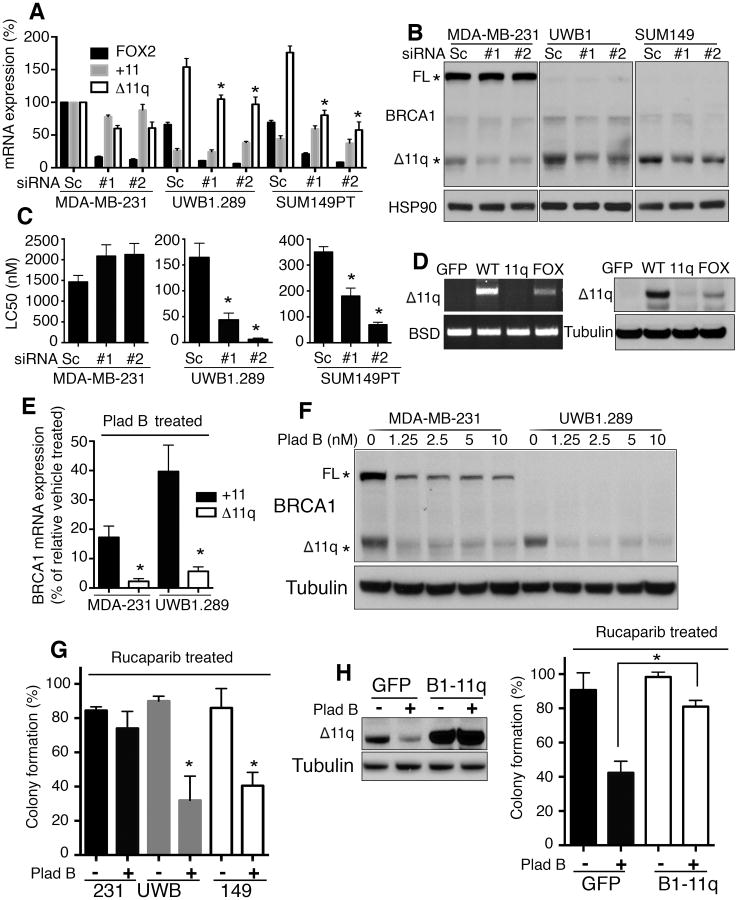
Breast and ovarian cancer patients harboring BRCA1/2 germline mutations have clinically benefitted from therapy with PARP inhibitor (PARPi) or platinum compounds, but acquired resistance limits clinical impact. In this study, we investigated the impact of mutations on BRCA1 isoform expression and therapeutic response. Cancer cell lines and tumors harboring mutations in exon 11 of BRCA1 express a BRCA1-Δ11q splice variant lacking the majority of exon 11. The introduction of frameshift mutations to exon 11 resulted in nonsense-mediated mRNA decay of full-length, but not the BRCA1-Δ11q isoform. CRISPR/Cas9 gene editing as well as overexpression experiments revealed that the BRCA1-Δ11q protein was capable of promoting partial PARPi and cisplatin resistance relative to full-length BRCA1, both in vitro and in vivo Furthermore, spliceosome inhibitors reduced BRCA1-Δ11q levels and sensitized cells carrying exon 11 mutations to PARPi treatment. Taken together, our results provided evidence that cancer cells employ a strategy to remove deleterious germline BRCA1 mutations through alternative mRNA splicing, giving rise to isoforms that retain residual activity and contribute to therapeutic resistance. Cancer Res; 76(9); 2778-90. ©2016 AACR.
©2016 American Association for Cancer Research.
Conflict of interest statement
Conflicts of interest: G.I.S. has served as an investigator in trials of rucaparib and Dana-Farber Cancer Institute has received funding from Clovis Oncology for the conduct of these trials. G.I.S. has also participated in advisory boards for Clovis. The remaining authors declare no conflicts of interest.
Figures
References
-
- Szabo CI, King MC. Inherited breast and ovarian cancer. Hum Mol Genet. 1995;4 Spec No:1811–1817. - PubMed
-
- Friedman LS, Ostermeyer EA, Szabo CI, Dowd P, Lynch ED, Rowell SE, King MC. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. 1994;8:399–404. - PubMed
-
- Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM, Mazoyer S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum Mol Genet. 2002;11:2805–2814. - PubMed
-
- Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. - PubMed
-
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
