

Spatial organization shapes the turnover of a bacterial transcriptome
- PMID: 27198188
- PMCID: PMC4874777
- DOI: 10.7554/eLife.13065
Spatial organization shapes the turnover of a bacterial transcriptome
Abstract
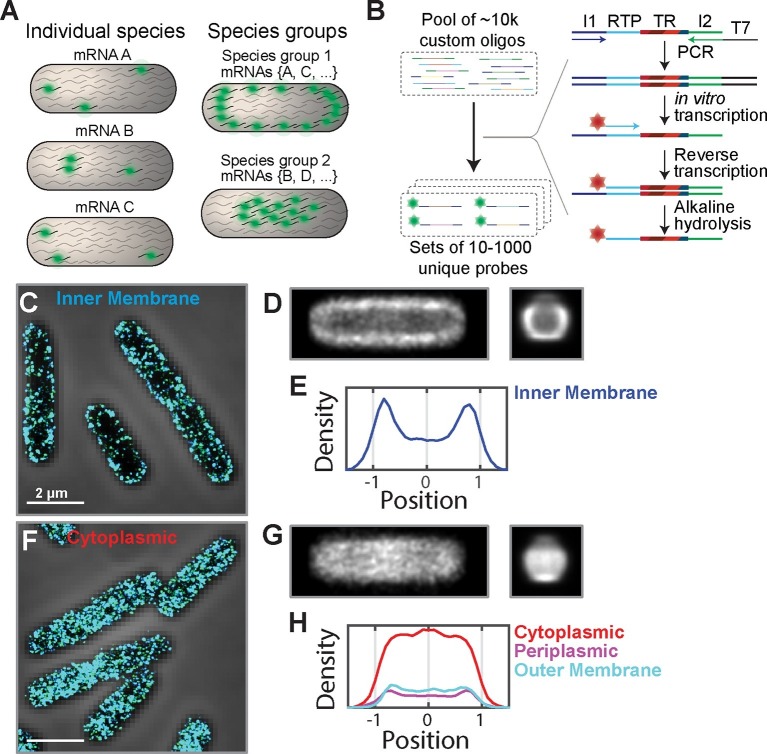
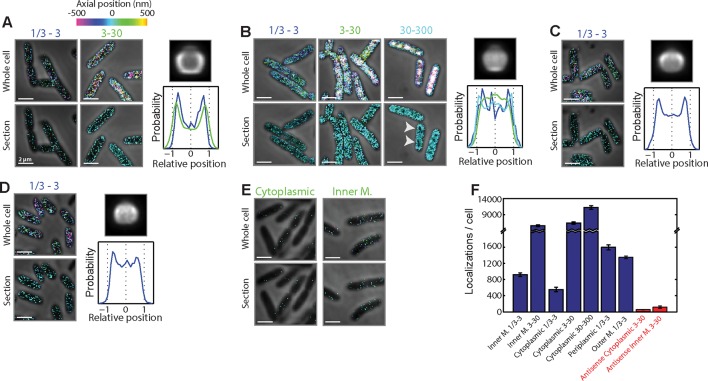
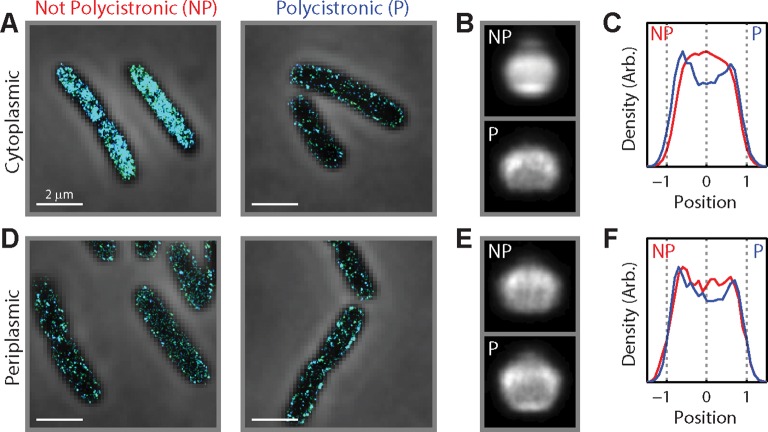
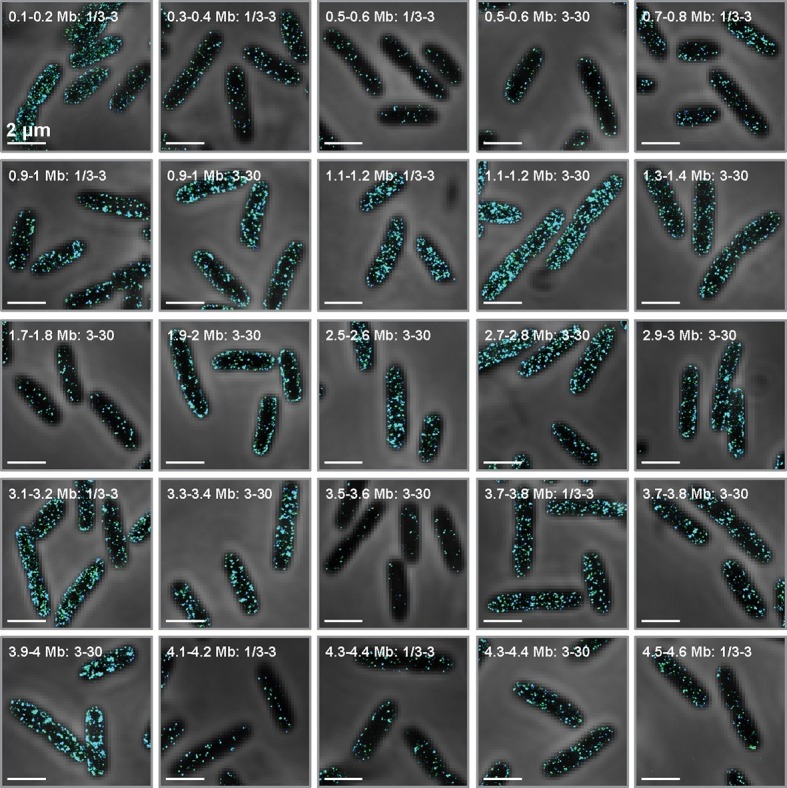
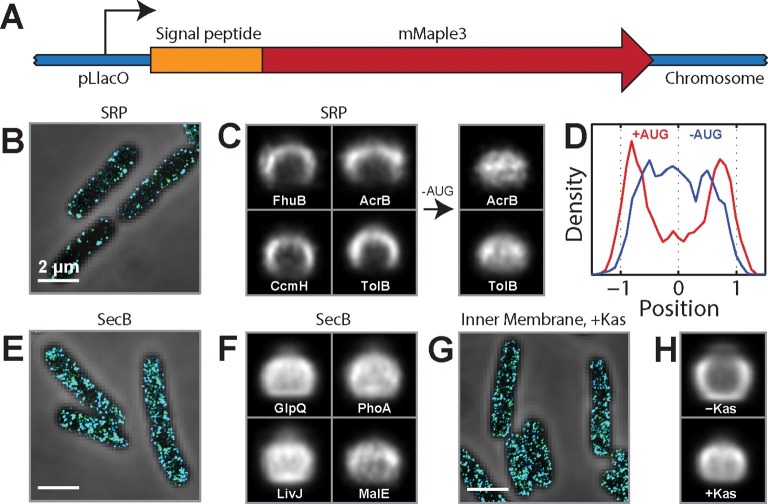
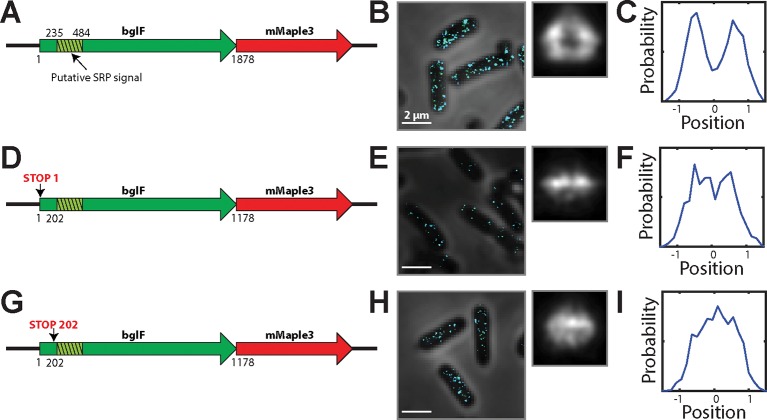
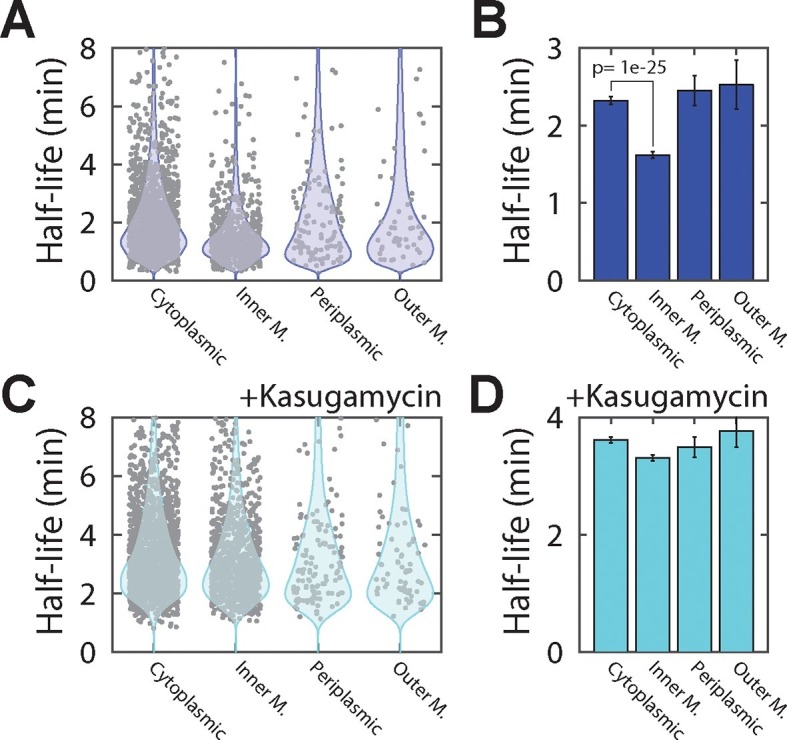
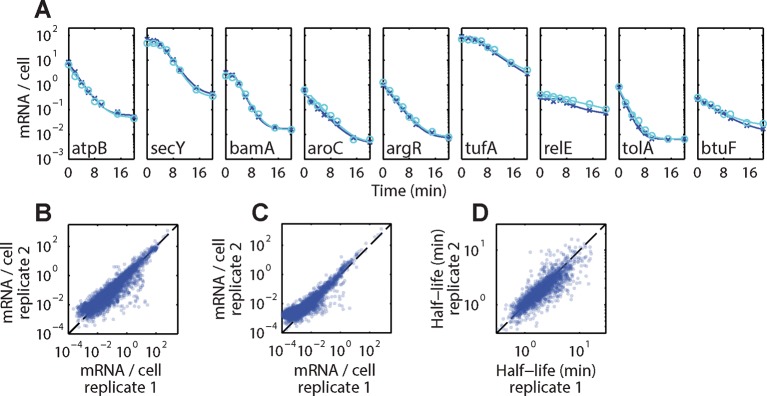
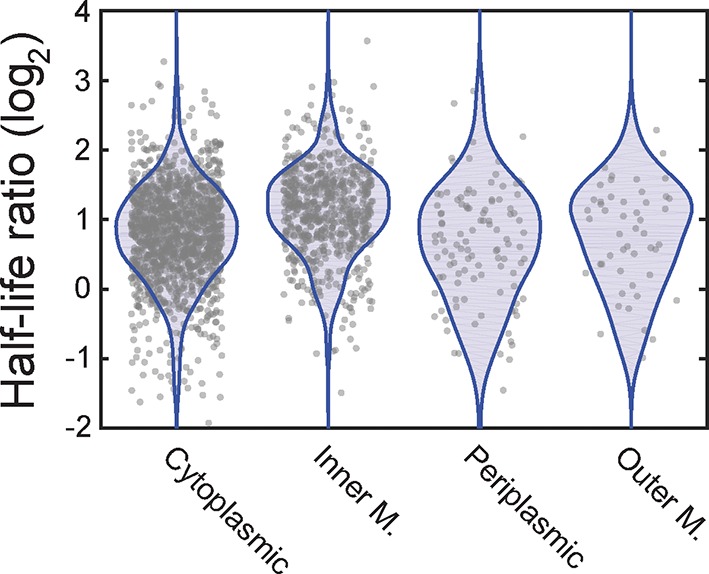
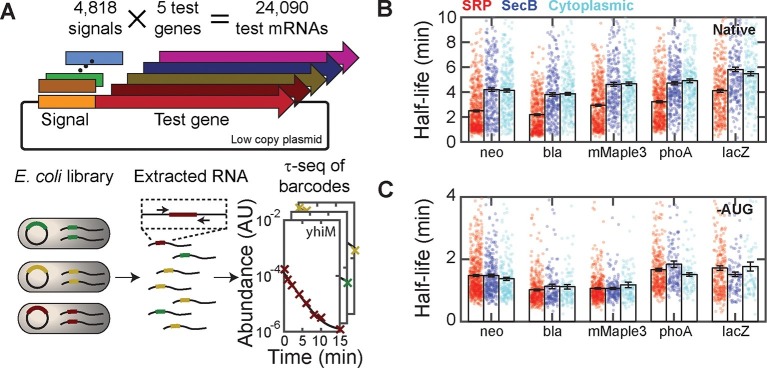
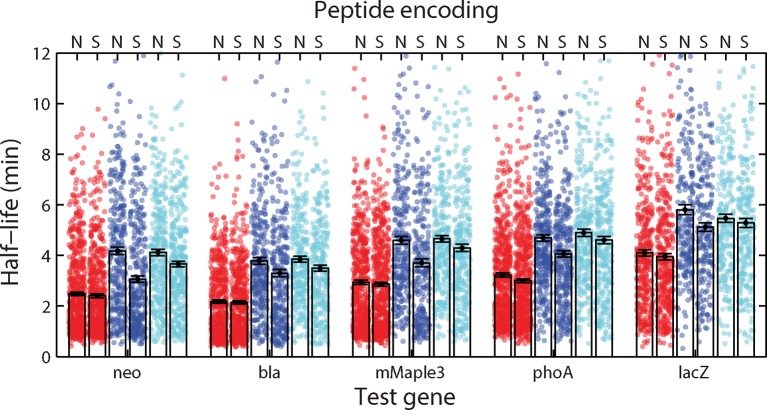
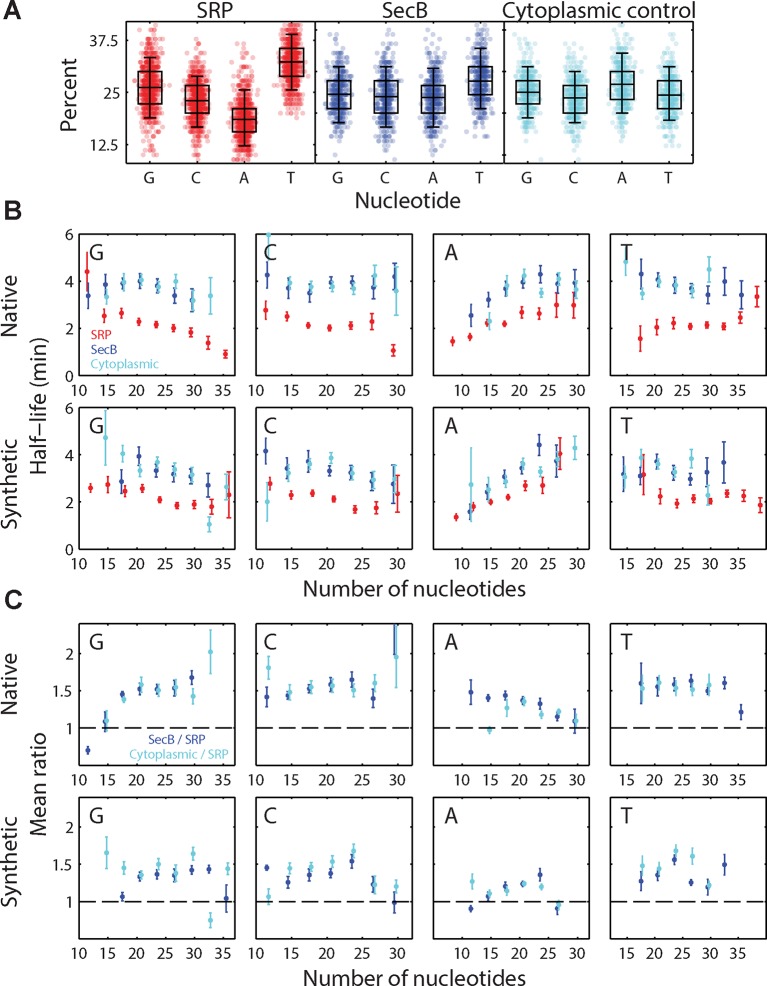
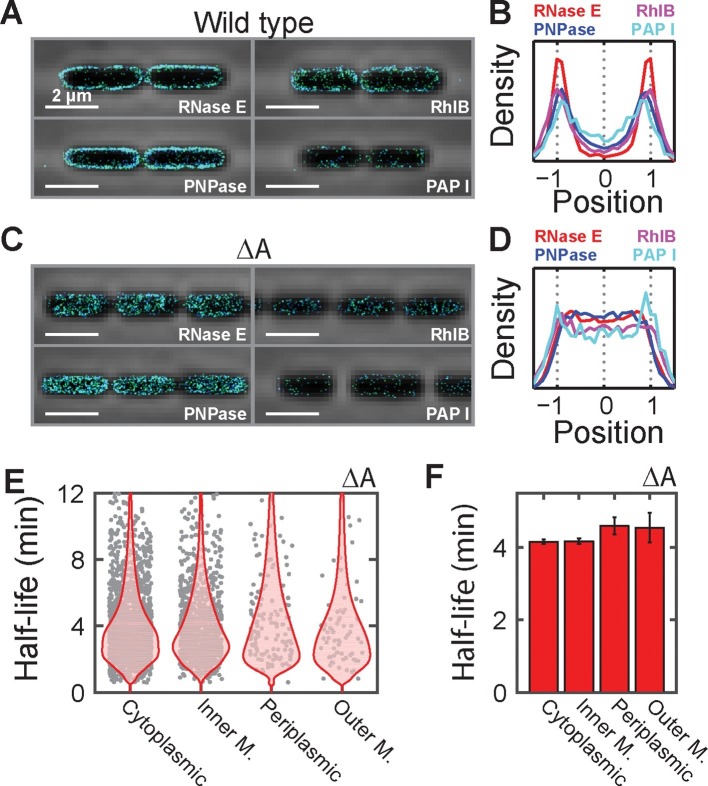
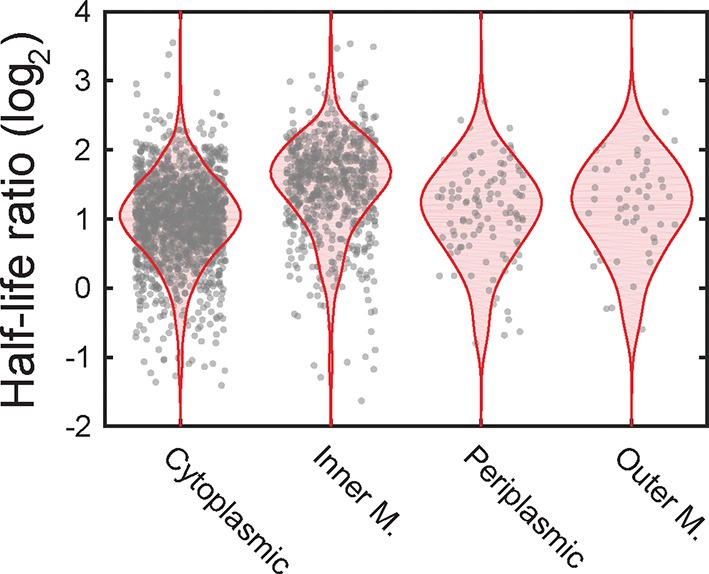
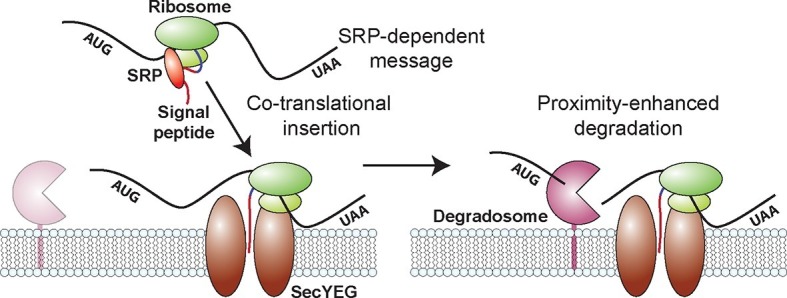
Spatial organization of the transcriptome has emerged as a powerful means for regulating the post-transcriptional fate of RNA in eukaryotes; however, whether prokaryotes use RNA spatial organization as a mechanism for post-transcriptional regulation remains unclear. Here we used super-resolution microscopy to image the E. coli transcriptome and observed a genome-wide spatial organization of RNA: mRNAs encoding inner-membrane proteins are enriched at the membrane, whereas mRNAs encoding outer-membrane, cytoplasmic and periplasmic proteins are distributed throughout the cytoplasm. Membrane enrichment is caused by co-translational insertion of signal peptides recognized by the signal-recognition particle. Time-resolved RNA-sequencing revealed that degradation rates of inner-membrane-protein mRNAs are on average greater that those of the other mRNAs and that this selective destabilization of inner-membrane-protein mRNAs is abolished by dissociating the RNA degradosome from the membrane. Together, these results demonstrate that the bacterial transcriptome is spatially organized and suggest that this organization shapes the post-transcriptional dynamics of mRNAs.
Keywords: E. coli; RNA degradation; biophysics; cell biology; fluorescence in situ hybridization; next-generation sequencing; spatial organization; structural biology; superresolution microscopy; transcriptome.
Conflict of interest statement
XZ: Reviewing editor,
The other authors declare that no competing interests exist.
Figures

Comment in
-
Finding FISH in a small pond.Elife. 2016 May 20;5:e16598. doi: 10.7554/eLife.16598. Elife. 2016. PMID: 27198190 Free PMC article.
-
Bacterial transcription: A tale of two specificities.Nat Rev Microbiol. 2016 Jul;14(7):404-5. doi: 10.1038/nrmicro.2016.88. Epub 2016 Jun 6. Nat Rev Microbiol. 2016. PMID: 27265767 No abstract available.
References
-
- Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, Chang Y, Li JB, Senaratne TN, Williams BR, Rouillard JM, Wu CT. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21301–21306. doi: 10.1073/pnas.1213818110. - DOI - PMC - PubMed
-
- Bernstein JA, Khodursky AB, Lin PH, Lin-Chao S, Cohen SN. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:9697–9702. doi: 10.1073/pnas.112318199. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
