

Reconstruction of gene regulatory networks reveals chromatin remodelers and key transcription factors in tumorigenesis
- PMID: 27198694
- PMCID: PMC4872343
- DOI: 10.1186/s13073-016-0310-3
Reconstruction of gene regulatory networks reveals chromatin remodelers and key transcription factors in tumorigenesis
Abstract
Background: Alterations in genetic and epigenetic landscapes are known to contribute to the development of different types of cancer. However, the mechanistic links between transcription factors and the epigenome which coordinate the deregulation of gene networks during cell transformation are largely unknown.
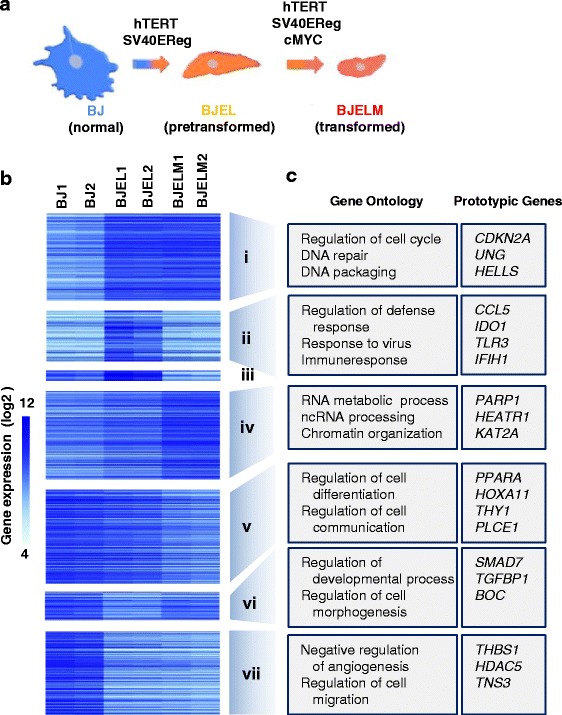
Methods: We used an isogenic model of stepwise tumorigenic transformation of human primary cells to monitor the progressive deregulation of gene networks upon immortalization and oncogene-induced transformation. We applied a systems biology approach by combining transcriptome and epigenome data for each step during transformation and integrated transcription factor-target gene associations in order to reconstruct the gene regulatory networks that are at the basis of the transformation process.
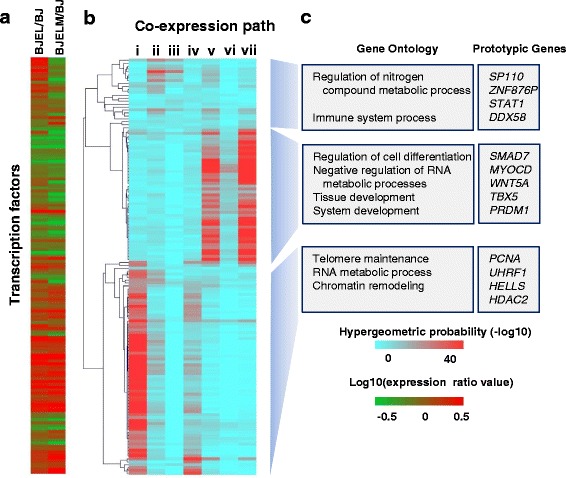
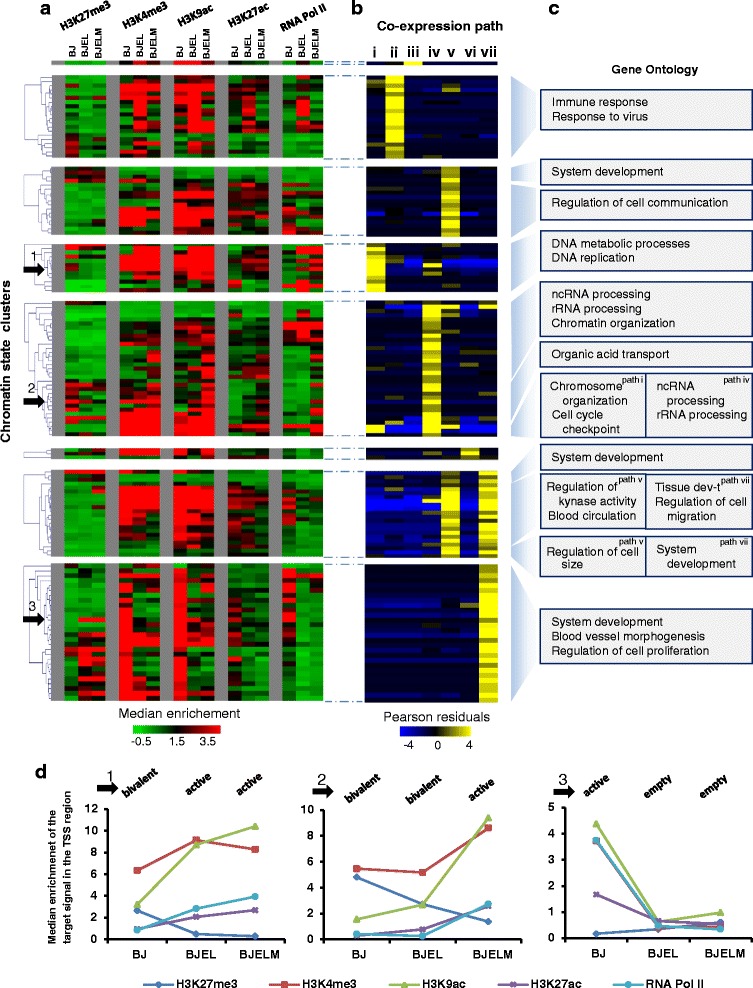
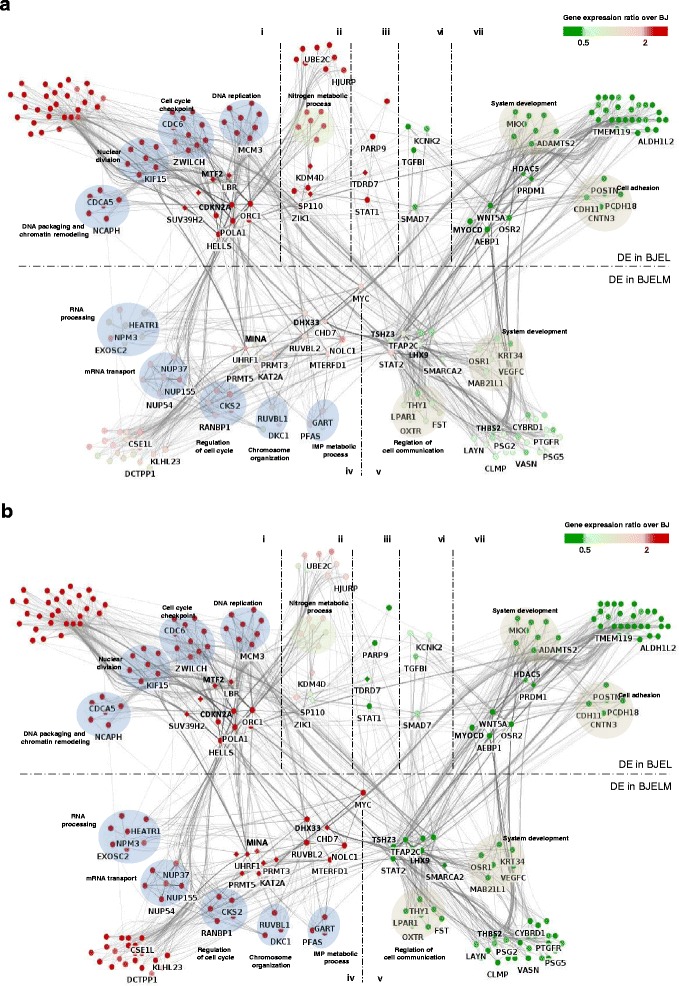
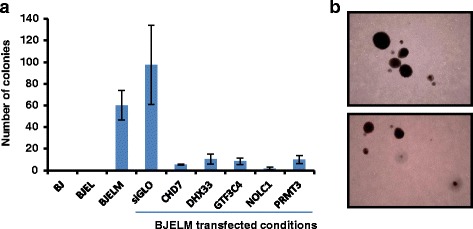
Results: We identified 142 transcription factors and 24 chromatin remodelers/modifiers (CRMs) which are preferentially associated with specific co-expression pathways that originate from deregulated gene programming during tumorigenesis. These transcription factors are involved in the regulation of divers processes, including cell differentiation, the immune response, and the establishment/modification of the epigenome. Unexpectedly, the analysis of chromatin state dynamics revealed patterns that distinguish groups of genes which are not only co-regulated but also functionally related. Decortication of transcription factor targets enabled us to define potential key regulators of cell transformation which are engaged in RNA metabolism and chromatin remodeling.
Conclusions: We reconstructed gene regulatory networks that reveal the alterations occurring during human cellular tumorigenesis. Using these networks we predicted and validated several transcription factors as key players for the establishment of tumorigenic traits of transformed cells. Our study suggests a direct implication of CRMs in oncogene-induced tumorigenesis and identifies new CRMs involved in this process. This is the first comprehensive view of the gene regulatory network that is altered during the process of stepwise human cellular tumorigenesis in a virtually isogenic system.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
