

MicroRNA-30 inhibits neointimal hyperplasia by targeting Ca(2+)/calmodulin-dependent protein kinase IIδ (CaMKIIδ)
- PMID: 27199283
- PMCID: PMC4873751
- DOI: 10.1038/srep26166
MicroRNA-30 inhibits neointimal hyperplasia by targeting Ca(2+)/calmodulin-dependent protein kinase IIδ (CaMKIIδ)
Abstract
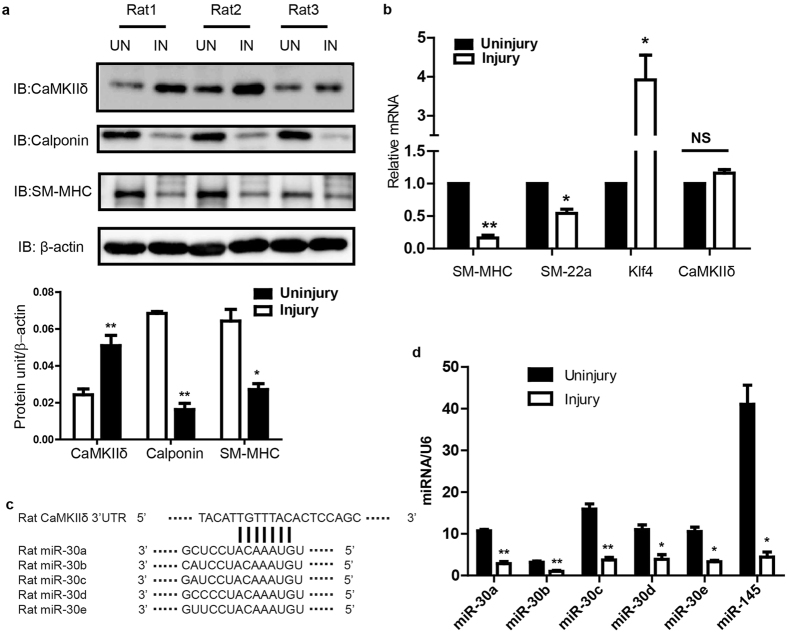
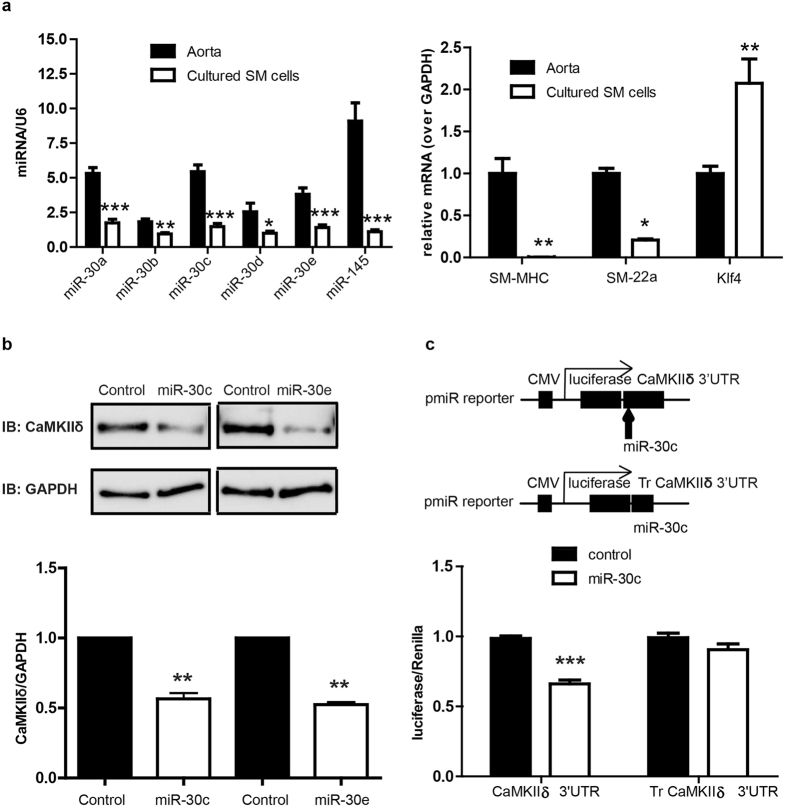
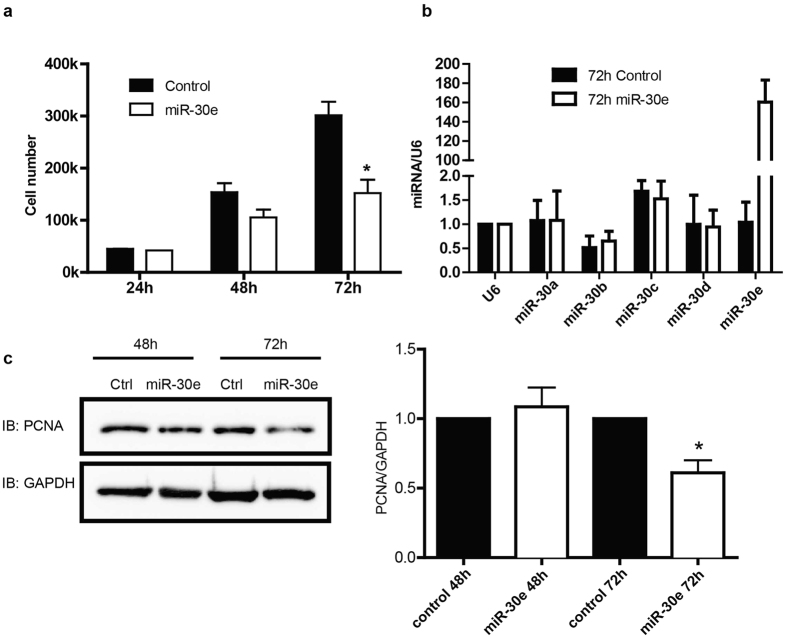
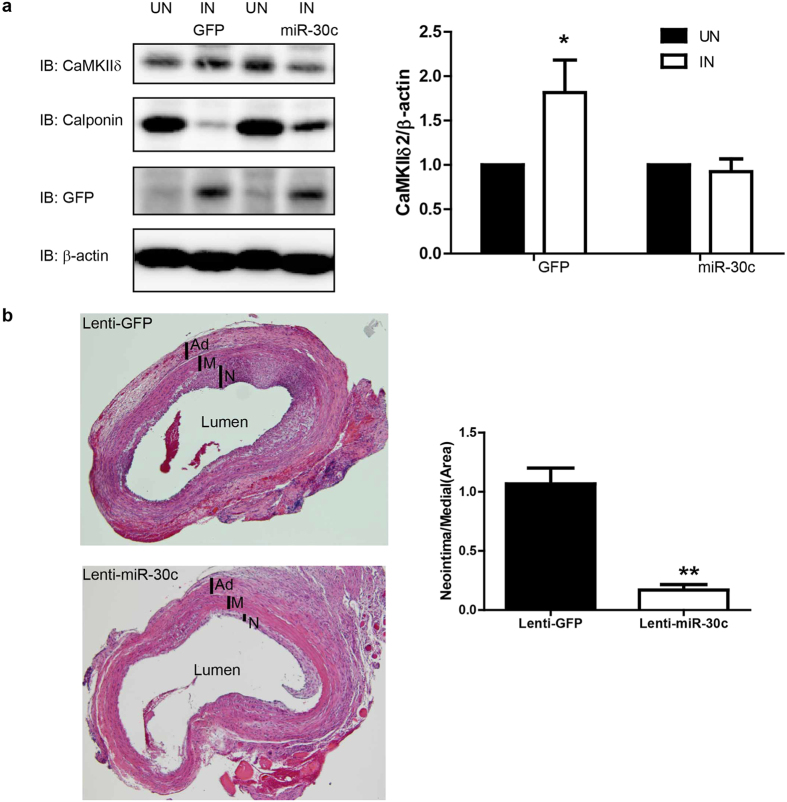
The multifunctional Ca(2+)/calmodulin-dependent protein kinase II δ-isoform (CaMKIIδ) promotes vascular smooth muscle (VSM) proliferation, migration, and injury-induced vascular wall neointima formation. The objective of this study was to test if microRNA-30 (miR-30) family members are endogenous regulators of CaMKIIδ expression following vascular injury and whether ectopic expression of miR-30 can inhibit CaMKIIδ-dependent VSM cell function and neointimal VSM hyperplasia induced by vascular injury. The CaMKIIδ 3'UTR contains a consensus miR-30 binding sequence that is highly conserved across species. A significant decrease in miR-30 family members and increase in CaMKIIδ2 protein expression, with no change in CaMKIIδ mRNA expression, was observed in medial layers of VSM 7 days post-injury. In vitro, overexpression of miR-30c or miR-30e inhibited CaMKIIδ2 protein expression by ~50% in cultured rat aortic VSM cells, and inhibited VSM cell proliferation and migration. In vivo, lenti-viral delivery of miR-30c into injured rat carotid arteries prevented the injury-induced increase in CaMKIIδ2. Furthermore, neointima formation was dramatically inhibited by lenti-viral delivery of miR-30c in the injured medial smooth muscle. These studies define a novel mechanism for regulating CaMKIIδ expression in VSM and provide a new potential therapeutic strategy to reduce progression of vascular proliferative diseases, including atherosclerosis and restenosis.
Figures

Similar articles
-
CaMKII-delta isoform regulation of neointima formation after vascular injury.Arterioscler Thromb Vasc Biol. 2008 Mar;28(3):441-7. doi: 10.1161/ATVBAHA.107.156810. Epub 2007 Dec 20. Arterioscler Thromb Vasc Biol. 2008. PMID: 18096823
-
Ca2+/calmodulin-dependent protein kinase II-γ (CaMKIIγ) negatively regulates vascular smooth muscle cell proliferation and vascular remodeling.FASEB J. 2016 Mar;30(3):1051-64. doi: 10.1096/fj.15-279158. Epub 2015 Nov 13. FASEB J. 2016. PMID: 26567004 Free PMC article.
-
Thymine DNA glycosylase is a key regulator of CaMKIIγ expression and vascular smooth muscle phenotype.Am J Physiol Heart Circ Physiol. 2019 Nov 1;317(5):H969-H980. doi: 10.1152/ajpheart.00146.2019. Epub 2019 Sep 13. Am J Physiol Heart Circ Physiol. 2019. PMID: 31518169 Free PMC article.
-
Ca2+/Calmodulin-Dependent Protein Kinase II in Vascular Smooth Muscle.Adv Pharmacol. 2017;78:171-202. doi: 10.1016/bs.apha.2016.08.003. Epub 2016 Oct 14. Adv Pharmacol. 2017. PMID: 28212797 Review.
-
Ca2+/calmodulin-dependent protein kinase II function in vascular remodelling.J Physiol. 2012 Mar 15;590(6):1349-56. doi: 10.1113/jphysiol.2011.222232. Epub 2011 Nov 28. J Physiol. 2012. PMID: 22124148 Free PMC article. Review.
Cited by
-
Expression of the microRNA-30 family in pulmonary arterial hypertension and the role of microRNA-30d-5p in the regulation of pulmonary arterial smooth muscle cell toxicity and apoptosis.Exp Ther Med. 2022 Jan;23(1):108. doi: 10.3892/etm.2021.11031. Epub 2021 Dec 2. Exp Ther Med. 2022. PMID: 34976150 Free PMC article.
-
MicroRNA-30e-5p promotes cell growth by targeting PTPN13 and indicates poor survival and recurrence in lung adenocarcinoma.J Cell Mol Med. 2017 Nov;21(11):2852-2862. doi: 10.1111/jcmm.13198. Epub 2017 Jun 27. J Cell Mol Med. 2017. PMID: 28653805 Free PMC article.
-
Role of CaMKII in diabetes induced vascular injury and its interaction with anti-diabetes therapy.Rev Endocr Metab Disord. 2024 Apr;25(2):369-382. doi: 10.1007/s11154-023-09855-9. Epub 2023 Dec 8. Rev Endocr Metab Disord. 2024. PMID: 38064002 Free PMC article. Review.
-
Evaluation of Arterial Histopathology and microRNA Expression That Underlie Ultrasonography Findings in Temporal Arteries of Patients with Giant Cell Arteritis.Int J Mol Sci. 2023 Jan 13;24(2):1572. doi: 10.3390/ijms24021572. Int J Mol Sci. 2023. PMID: 36675088 Free PMC article.
-
Role of miRNA in the Regulatory Mechanisms of Estrogens in Cardiovascular Ageing.Oxid Med Cell Longev. 2018 Dec 20;2018:6082387. doi: 10.1155/2018/6082387. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 30671171 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
