

Nipah Virus C Protein Recruits Tsg101 to Promote the Efficient Release of Virus in an ESCRT-Dependent Pathway
- PMID: 27203423
- PMCID: PMC4874542
- DOI: 10.1371/journal.ppat.1005659
Nipah Virus C Protein Recruits Tsg101 to Promote the Efficient Release of Virus in an ESCRT-Dependent Pathway
Abstract
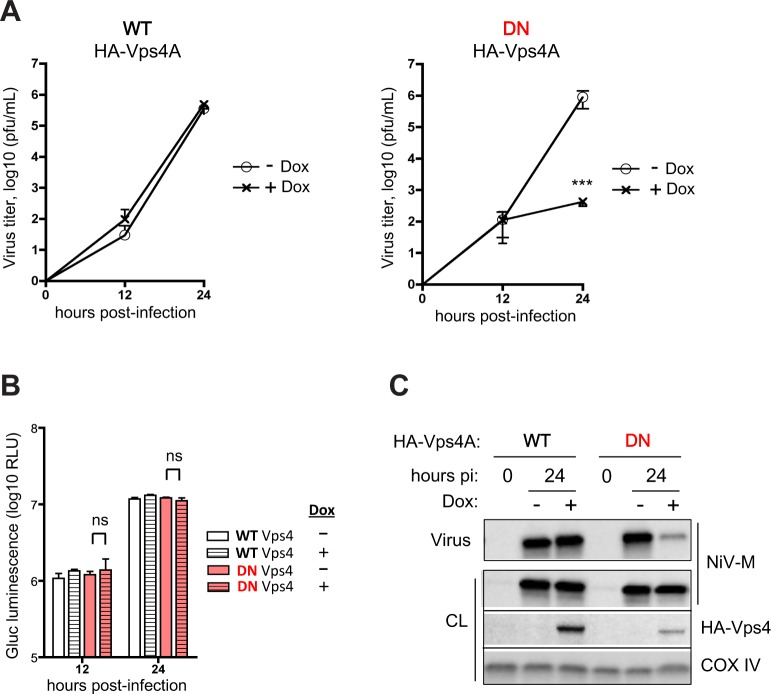
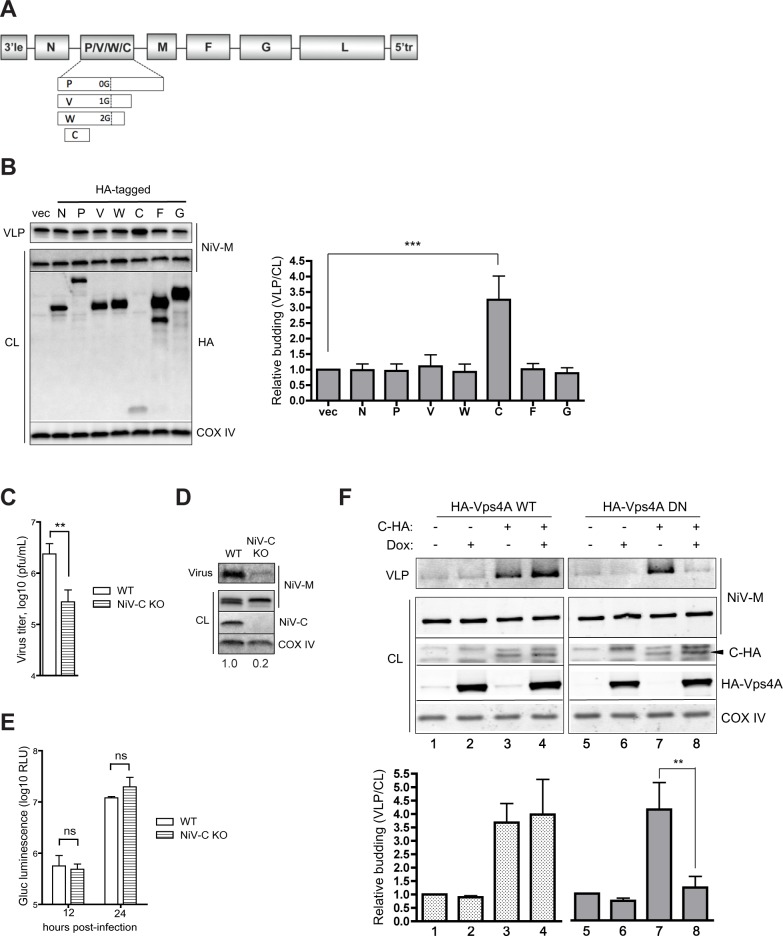
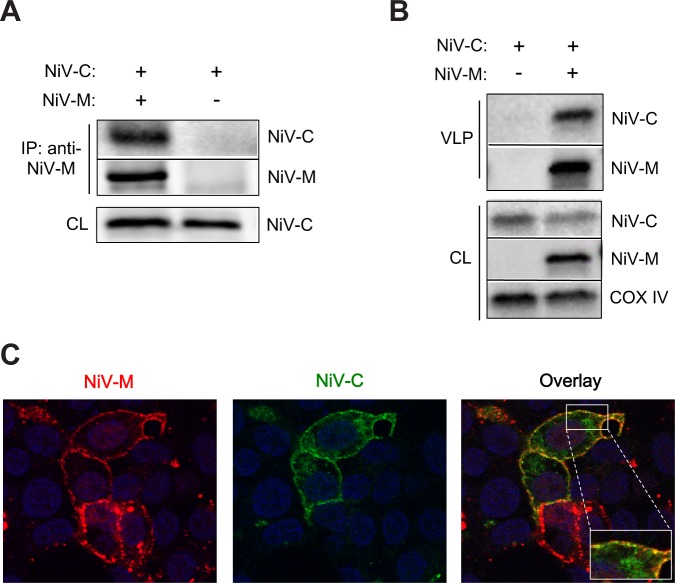
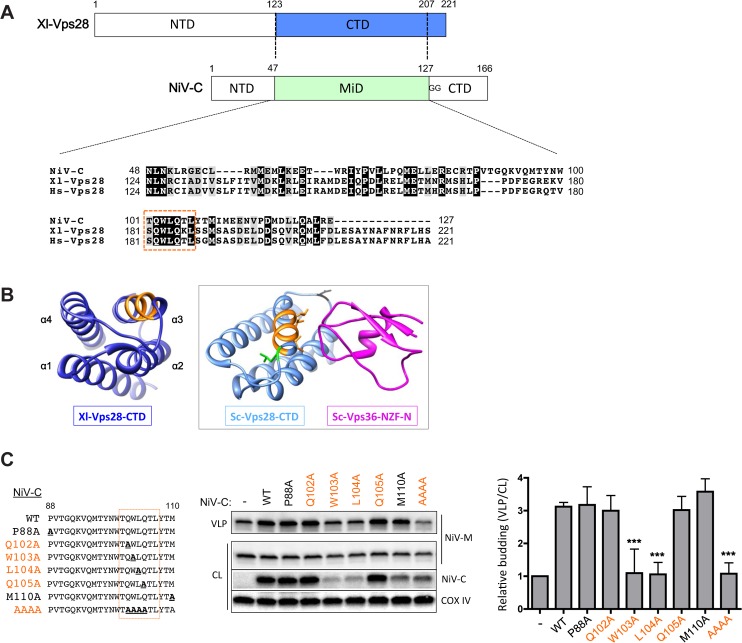
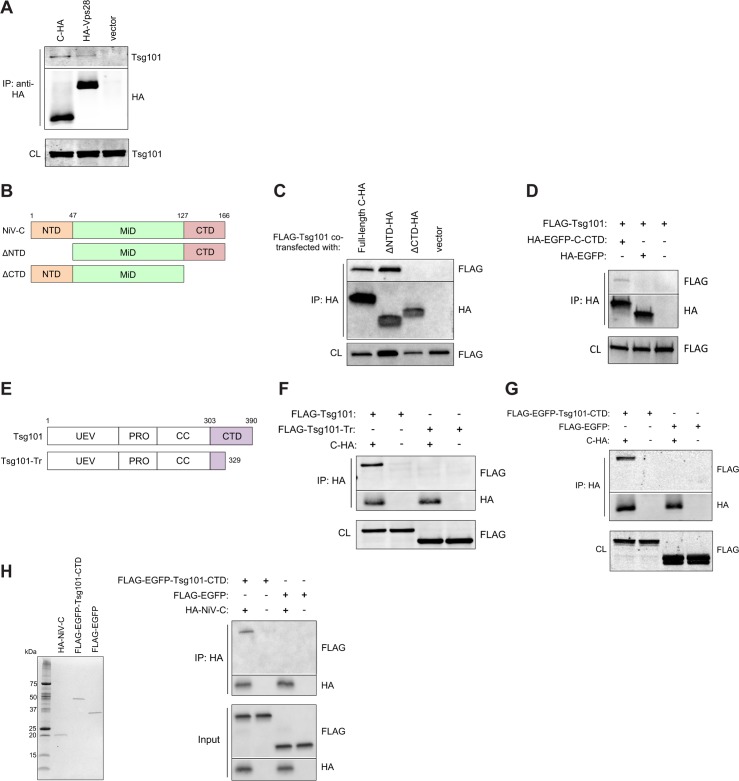
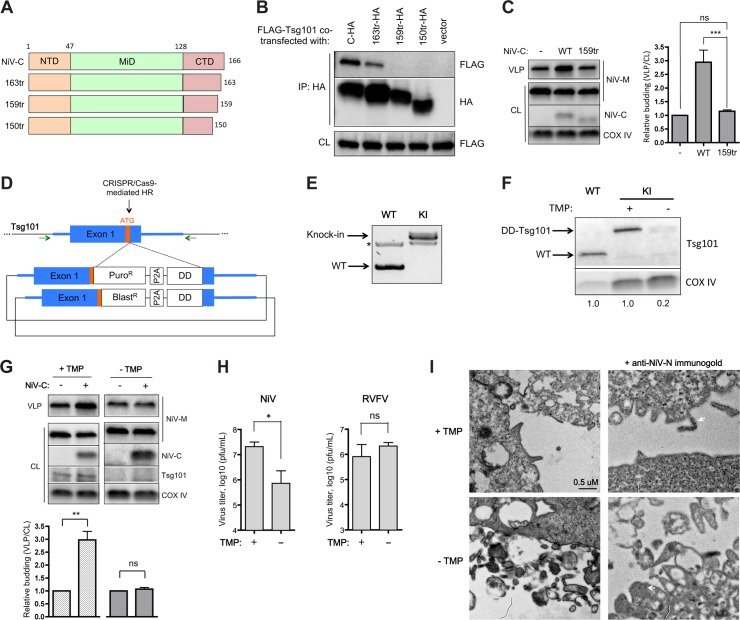
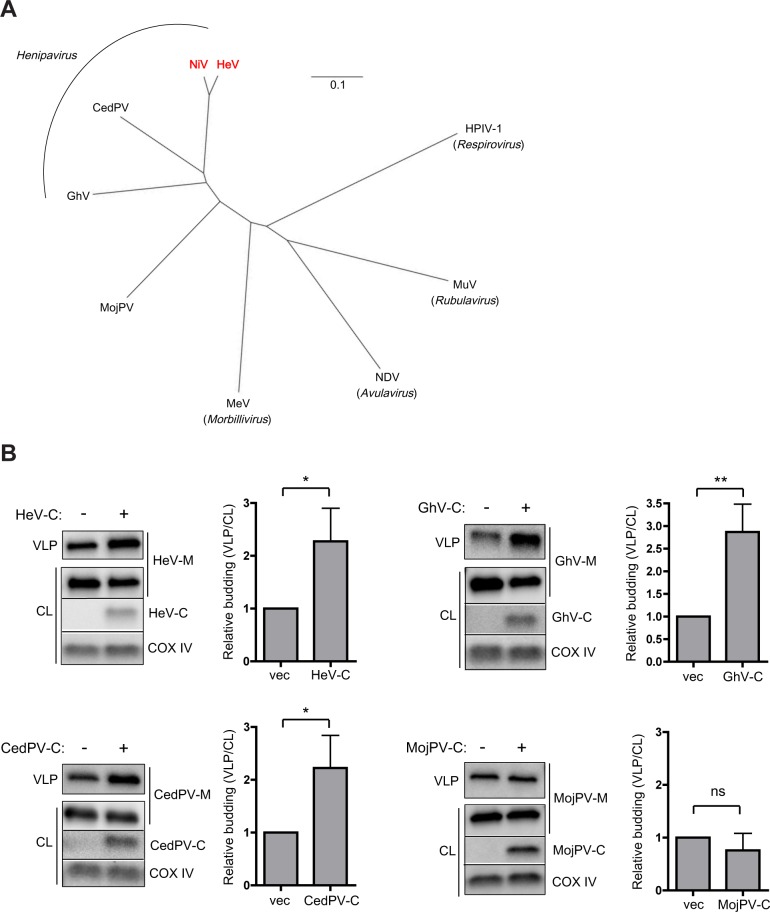
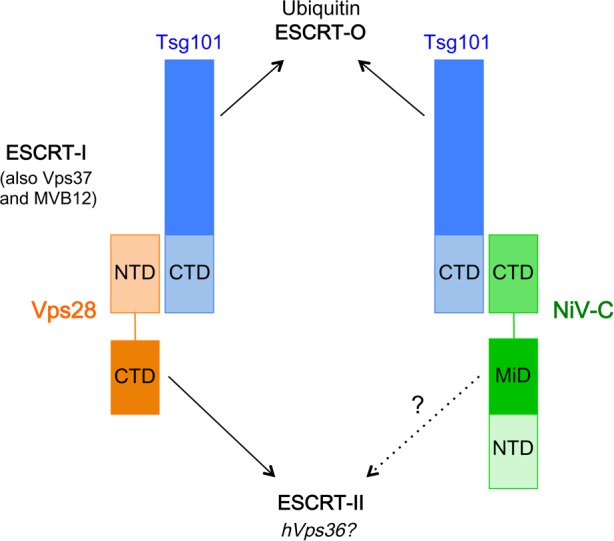
The budding of Nipah virus, a deadly member of the Henipavirus genus within the Paramyxoviridae, has been thought to be independent of the host ESCRT pathway, which is critical for the budding of many enveloped viruses. This conclusion was based on the budding properties of the virus matrix protein in the absence of other virus components. Here, we find that the virus C protein, which was previously investigated for its role in antagonism of innate immunity, recruits the ESCRT pathway to promote efficient virus release. Inhibition of ESCRT or depletion of the ESCRT factor Tsg101 abrogates the C enhancement of matrix budding and impairs live Nipah virus release. Further, despite the low sequence homology of the C proteins of known henipaviruses, they all enhance the budding of their cognate matrix proteins, suggesting a conserved and previously unknown function for the henipavirus C proteins.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Interaction with Tsg101 is necessary for the efficient transport and release of nucleocapsids in marburg virus-infected cells.PLoS Pathog. 2014 Oct 16;10(10):e1004463. doi: 10.1371/journal.ppat.1004463. eCollection 2014 Oct. PLoS Pathog. 2014. PMID: 25330247 Free PMC article.
-
HIV-1 Vpr Abrogates the Effect of TSG101 Overexpression to Support Virus Release.PLoS One. 2016 Sep 20;11(9):e0163100. doi: 10.1371/journal.pone.0163100. eCollection 2016. PLoS One. 2016. PMID: 27648839 Free PMC article.
-
Tumour susceptibility gene 101 and the vacuolar protein sorting pathway are required for the release of hepatitis E virions.J Gen Virol. 2011 Dec;92(Pt 12):2838-2848. doi: 10.1099/vir.0.035378-0. Epub 2011 Aug 31. J Gen Virol. 2011. PMID: 21880841
-
The regulation of Endosomal Sorting Complex Required for Transport and accessory proteins in multivesicular body sorting and enveloped viral budding - An overview.Int J Biol Macromol. 2019 Apr 15;127:1-11. doi: 10.1016/j.ijbiomac.2019.01.015. Epub 2019 Jan 4. Int J Biol Macromol. 2019. PMID: 30615963 Review.
-
Virus budding and the ESCRT pathway.Cell Host Microbe. 2013 Sep 11;14(3):232-41. doi: 10.1016/j.chom.2013.08.012. Cell Host Microbe. 2013. PMID: 24034610 Free PMC article. Review.
Cited by
-
The nanoscale organization of Nipah virus matrix protein revealed by super-resolution microscopy.Biophys J. 2022 Jun 21;121(12):2290-2296. doi: 10.1016/j.bpj.2022.05.026. Epub 2022 May 25. Biophys J. 2022. PMID: 35614854 Free PMC article.
-
Extracellular Vesicles and Ebola Virus: A New Mechanism of Immune Evasion.Viruses. 2019 May 2;11(5):410. doi: 10.3390/v11050410. Viruses. 2019. PMID: 31052499 Free PMC article. Review.
-
Morphogenesis of Bullet-Shaped Rabies Virus Particles Regulated by TSG101.J Virol. 2023 May 31;97(5):e0043823. doi: 10.1128/jvi.00438-23. Epub 2023 Apr 12. J Virol. 2023. PMID: 37042780 Free PMC article.
-
ESCRT-I Protein Tsg101 Plays a Role in the Post-macropinocytic Trafficking and Infection of Endothelial Cells by Kaposi's Sarcoma-Associated Herpesvirus.PLoS Pathog. 2016 Oct 20;12(10):e1005960. doi: 10.1371/journal.ppat.1005960. eCollection 2016 Oct. PLoS Pathog. 2016. PMID: 27764233 Free PMC article.
-
Orthoparamyxovirinae C Proteins Have a Common Origin and a Common Structural Organization.Biomolecules. 2023 Mar 1;13(3):455. doi: 10.3390/biom13030455. Biomolecules. 2023. PMID: 36979390 Free PMC article.
References
-
- Demirov DG, Freed EO (2004) Retrovirus budding. Virus Res 106: 87–102. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
