

Endocrine neoplasms in familial syndromes of hyperparathyroidism
- PMID: 27207564
- PMCID: PMC4917437
- DOI: 10.1530/ERC-16-0059
Endocrine neoplasms in familial syndromes of hyperparathyroidism
Abstract
Familial syndromes of hyperparathyroidism, including multiple endocrine neoplasia type 1 (MEN1), multiple endocrine neoplasia type 2A (MEN2A), and the hyperparathyroidism-jaw tumor (HPT-JT), comprise 2-5% of primary hyperparathyroidism cases. Familial syndromes of hyperparathyroidism are also associated with a range of endocrine and nonendocrine tumors, including potential malignancies. Complications of the associated neoplasms are the major causes of morbidities and mortalities in these familial syndromes, e.g., parathyroid carcinoma in HPT-JT syndrome; thymic, bronchial, and enteropancreatic neuroendocrine tumors in MEN1; and medullary thyroid cancer and pheochromocytoma in MEN2A. Because of the different underlying mechanisms of neoplasia, these familial tumors may have different characteristics compared with their sporadic counterparts. Large-scale clinical trials are frequently lacking due to the rarity of these diseases. With technological advances and the development of new medications, the natural history, diagnosis, and management of these syndromes are also evolving. In this article, we summarize the recent knowledge on endocrine neoplasms in three familial hyperparathyroidism syndromes, with an emphasis on disease characteristics, molecular pathogenesis, recent developments in biochemical and radiological evaluation, and expert opinions on surgical and medical therapies. Because these familial hyperparathyroidism syndromes are associated with a wide variety of tumors in different organs, this review is focused on those endocrine neoplasms with malignant potential.
Keywords: hyperparathyroidism-jaw tumor (HPT-JT); malignant tumor; multiple endocrine neoplasia type 1 (MEN1); multiple endocrine neoplasia type 2A (MEN2A); neuroendocrine tumor.
© 2016 Society for Endocrinology.
Conflict of interest statement
Figures
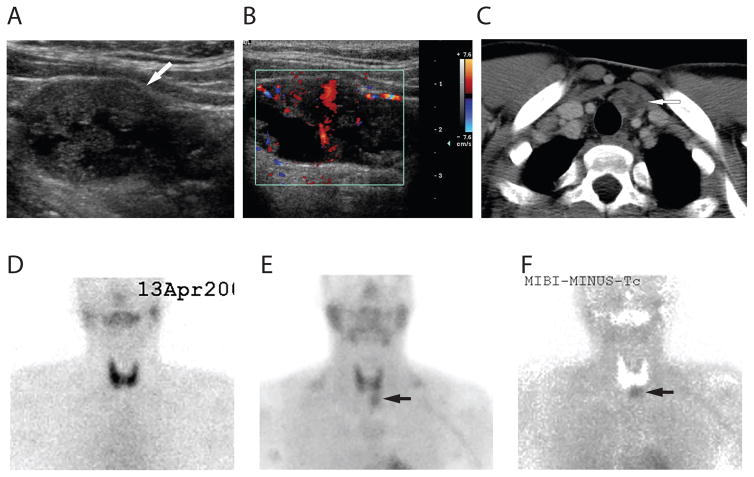
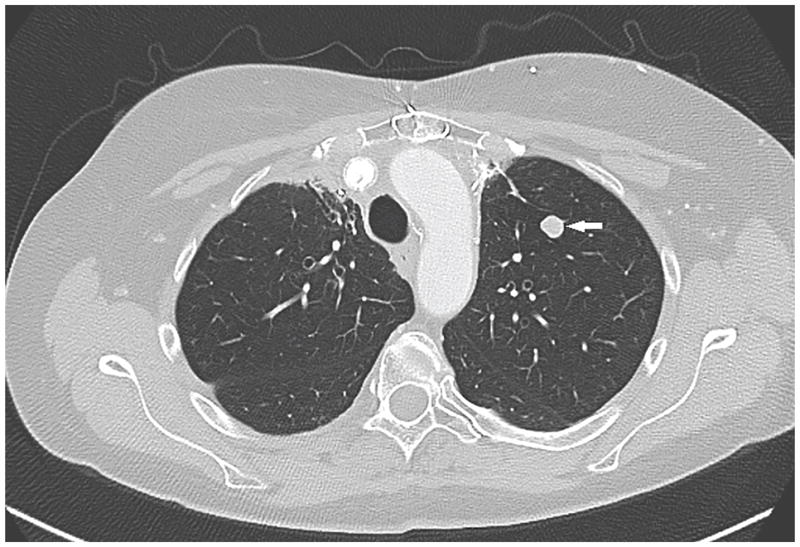
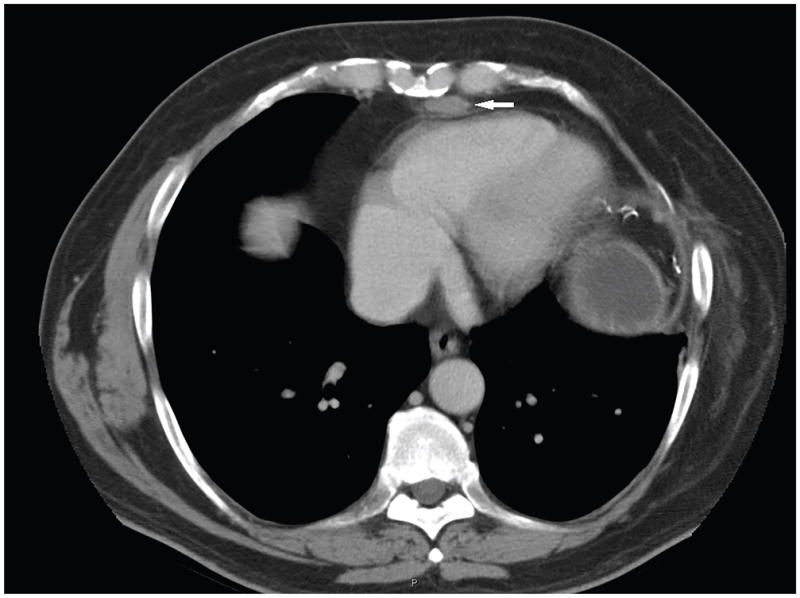
References
-
- Abe T, Sato M, Okumura T, Shioyama Y, Kiyoshima M, Asato Y, Saito H, Iijima T, Amemiya R, Nagai H. FDG PET/CT findings of thymic carcinoid and bronchial carcinoid in a patient with multiple neuroendocrine neoplasia type1. Clin Nucl Med. 2008;33:778–779. - PubMed
-
- Altemir Trallero J, Armengod Grao L, Aguillo Gutierrez E, Cabrejas Gomez C, Ocon Breton J, Garcia Garcia B. Thymic carcinoid in the setting of a multiple endocrine neoplasia syndrome (MEN 1). Prophylactic thymectomy? Endocrinol Nutr. 2012;59:142–144. - PubMed
-
- Amano H, Yamada T, Jujoh T, Kuroda F, Sakao S, Tada Y, Kurosu K, Kasahara Y, Tanabe N, Takiguchi Y, et al. Case of thymic carcinoid associated with multiple endocrine neoplasia type I treated effectively with chemotherapy. Nihon Kokyuki Gakkai Zasshi. 2010;48:855–859. - PubMed
-
- Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic ultrasound is highly accurate and directs management in patients with neuroendocrine tumors of the pancreas. Am J Gastroenterol. 2000;95:2271–2277. - PubMed
-
- Asare EA, Sturgeon C, Winchester DJ, Liu L, Palis B, Perrier ND, Evans DB, Winchester DP, Wang TS. Parathyroid Carcinoma: An Update on Treatment Outcomes and Prognostic Factors from the National Cancer Data Base (NCDB) Ann Surg Oncol. 2015;22:3990–3995. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
