

A Homolog of Blade-On-Petiole 1 and 2 (BOP1/2) Controls Internode Length and Homeotic Changes of the Barley Inflorescence
- PMID: 27208226
- PMCID: PMC4902598
- DOI: 10.1104/pp.16.00124
A Homolog of Blade-On-Petiole 1 and 2 (BOP1/2) Controls Internode Length and Homeotic Changes of the Barley Inflorescence
Abstract
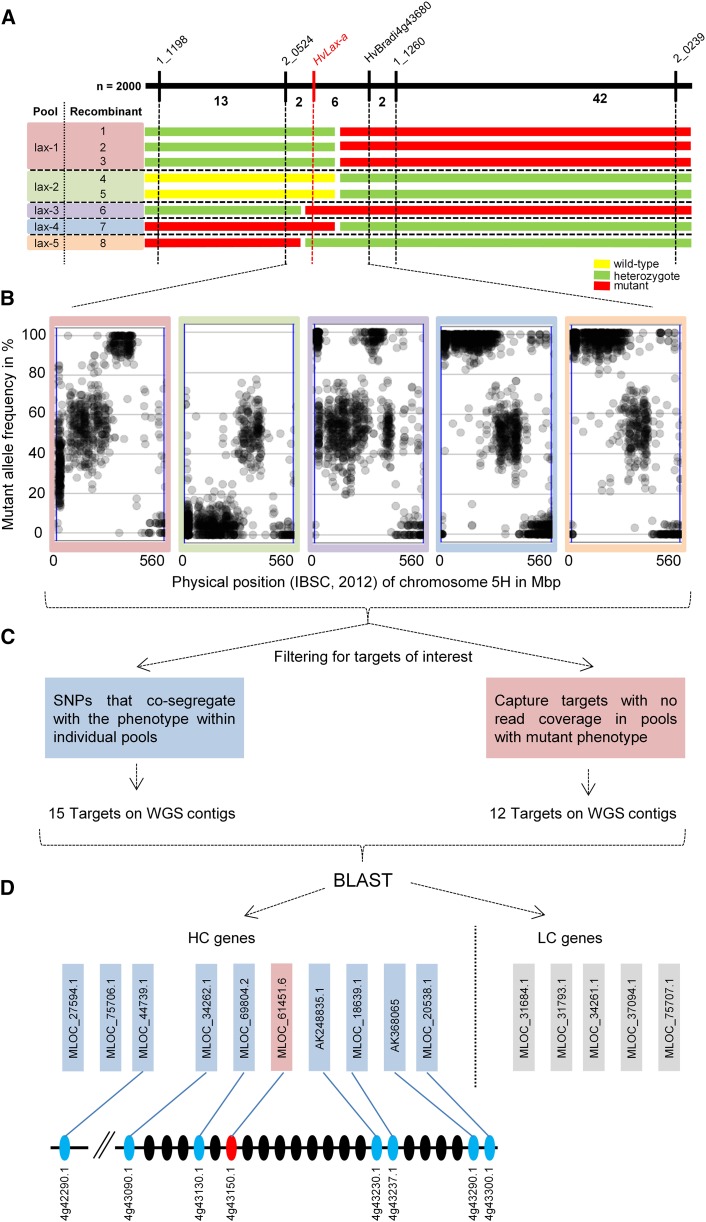
Inflorescence architecture in small-grain cereals has a direct effect on yield and is an important selection target in breeding for yield improvement. We analyzed the recessive mutation laxatum-a (lax-a) in barley (Hordeum vulgare), which causes pleiotropic changes in spike development, resulting in (1) extended rachis internodes conferring a more relaxed inflorescence, (2) broadened base of the lemma awns, (3) thinner grains that are largely exposed due to reduced marginal growth of the palea and lemma, and (4) and homeotic conversion of lodicules into two stamenoid structures. Map-based cloning enforced by mapping-by-sequencing of the mutant lax-a locus enabled the identification of a homolog of BLADE-ON-PETIOLE1 (BOP1) and BOP2 as the causal gene. Interestingly, the recently identified barley uniculme4 gene also is a BOP1/2 homolog and has been shown to regulate tillering and leaf sheath development. While the Arabidopsis (Arabidopsis thaliana) BOP1 and BOP2 genes act redundantly, the barley genes contribute independent effects in specifying the developmental growth of vegetative and reproductive organs, respectively. Analysis of natural genetic diversity revealed strikingly different haplotype diversity for the two paralogous barley genes, likely affected by the respective genomic environments, since no indication for an active selection process was detected.
© 2016 American Society of Plant Biologists. All Rights Reserved.
Figures



References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 - PubMed
-
- Andrés F, Romera-Branchat M, Martínez-Gallegos R, Patel V, Schneeberger K, Jang S, Altmüller J, Nürnberg P, Coupland G (2015) Floral induction in Arabidopsis by FLOWERING LOCUS T requires direct repression of BLADE-ON-PETIOLE genes by the homeodomain protein PENNYWISE. Plant Physiol 169: 2187–2199 - PMC - PubMed
-
- Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48 - PubMed
-
- Begun DJ, Aquadro CF (1992) Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
