

Eye on the B-ALL: B-cell receptor repertoires reveal persistence of numerous B-lymphoblastic leukemia subclones from diagnosis to relapse
- PMID: 27211266
- PMCID: PMC5155029
- DOI: 10.1038/leu.2016.142
Eye on the B-ALL: B-cell receptor repertoires reveal persistence of numerous B-lymphoblastic leukemia subclones from diagnosis to relapse
Abstract
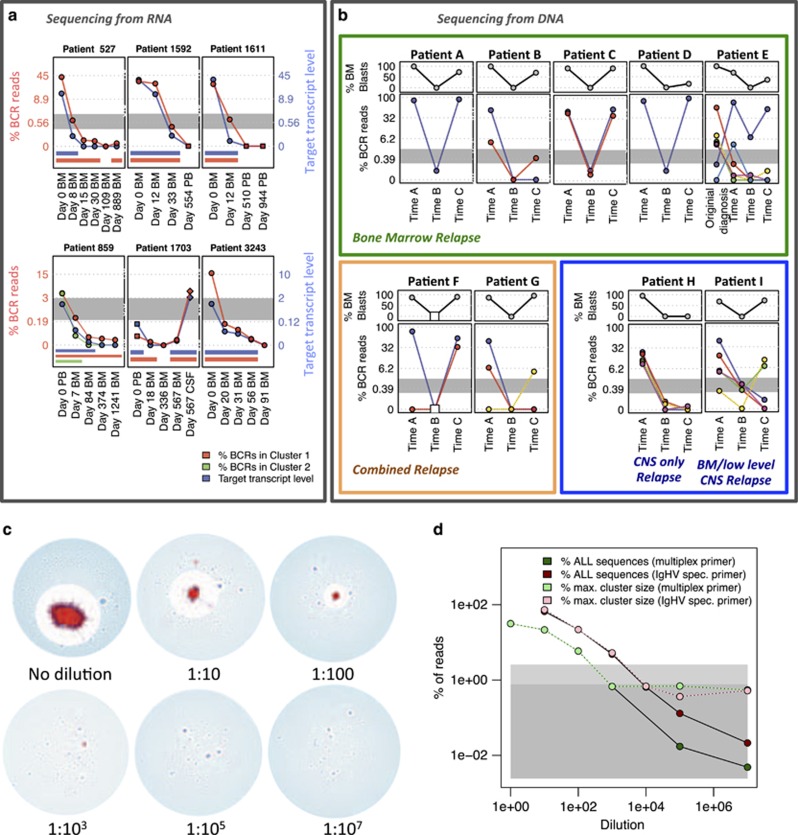
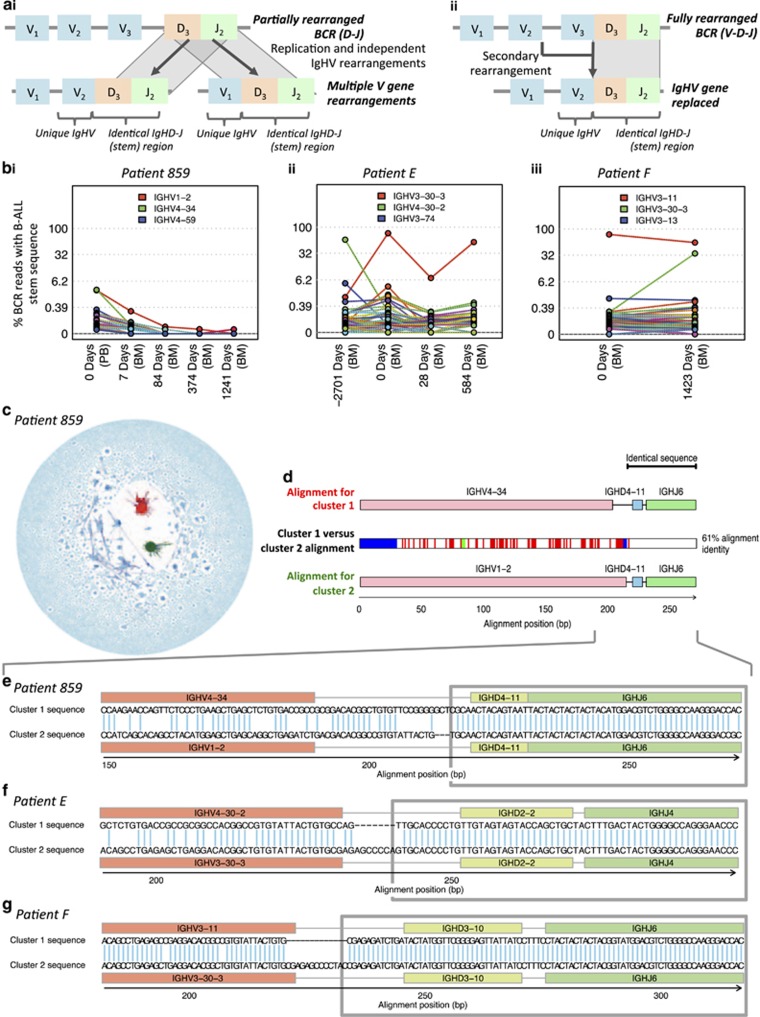
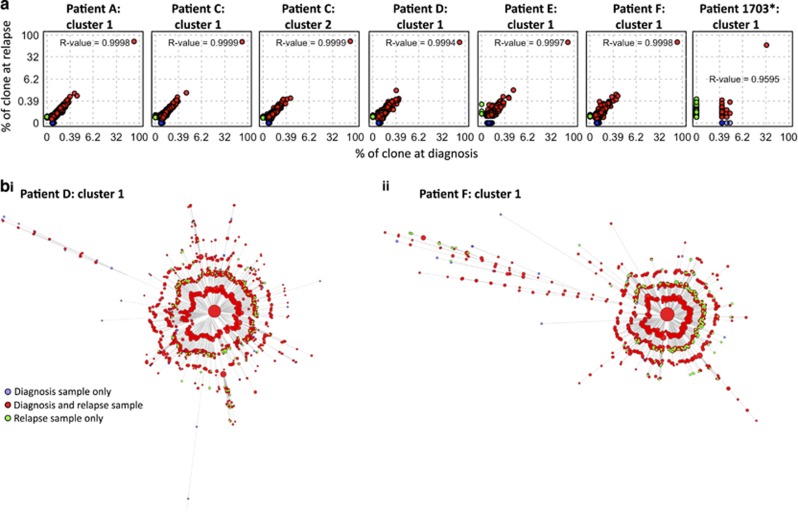
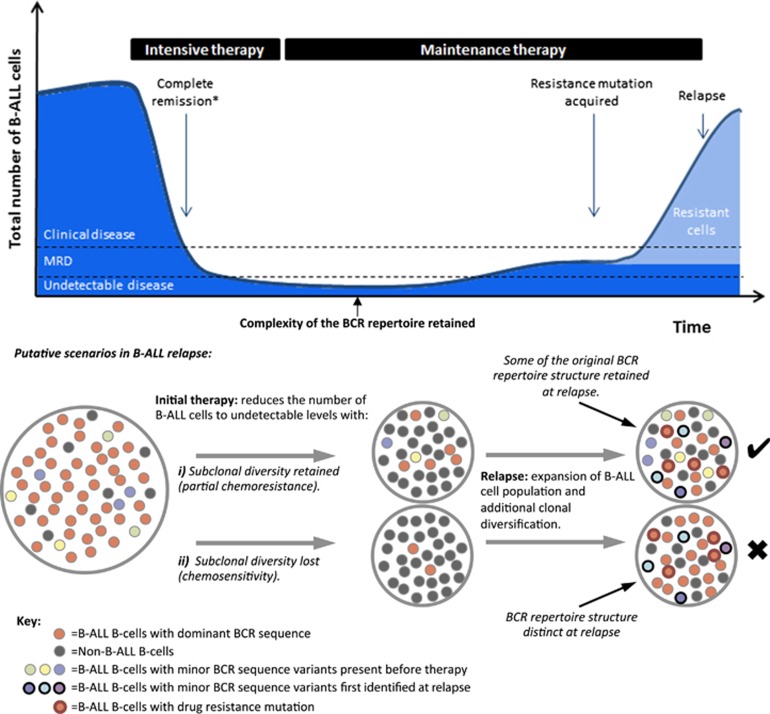
The strongest predictor of relapse in B-cell acute lymphoblastic leukemia (B-ALL) is the level of persistence of tumor cells after initial therapy. The high mutation rate of the B-cell receptor (BCR) locus allows high-resolution tracking of the architecture, evolution and clonal dynamics of B-ALL. Using longitudinal BCR repertoire sequencing, we find that the BCR undergoes an unexpectedly high level of clonal diversification in B-ALL cells through both somatic hypermutation and secondary rearrangements, which can be used for tracking the subclonal composition of the disease and detect minimal residual disease with unprecedented sensitivity. We go on to investigate clonal dynamics of B-ALL using BCR phylogenetic analyses of paired diagnosis-relapse samples and find that large numbers of small leukemic subclones present at diagnosis re-emerge at relapse alongside a dominant clone. Our findings suggest that in all informative relapsed patients, the survival of large numbers of clonogenic cells beyond initial chemotherapy is a surrogate for inherent partial chemoresistance or inadequate therapy, providing an increased opportunity for subsequent emergence of fully resistant clones. These results frame early cytoreduction as an important determinant of long-term outcome.
Figures
References
-
- Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet 2008; 371: 1030–1043. - PubMed
-
- Fielding AK. The treatment of adults with acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program 2008, 381–389, doi:10.1182/asheducation-2008.1.381. - PubMed
-
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 2006; 24: 541–570. - PubMed
-
- Maizels N. Somatic hypermutation: how many mechanisms diversify V region sequences? Cell 1995; 83: 9–12. - PubMed
-
- Kelsoe G. In situ studies of the germinal center reaction. Adv Immunol 1995; 60: 267–288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
