

Complement in disease: a defence system turning offensive
- PMID: 27211870
- PMCID: PMC4974115
- DOI: 10.1038/nrneph.2016.70
Complement in disease: a defence system turning offensive
Abstract
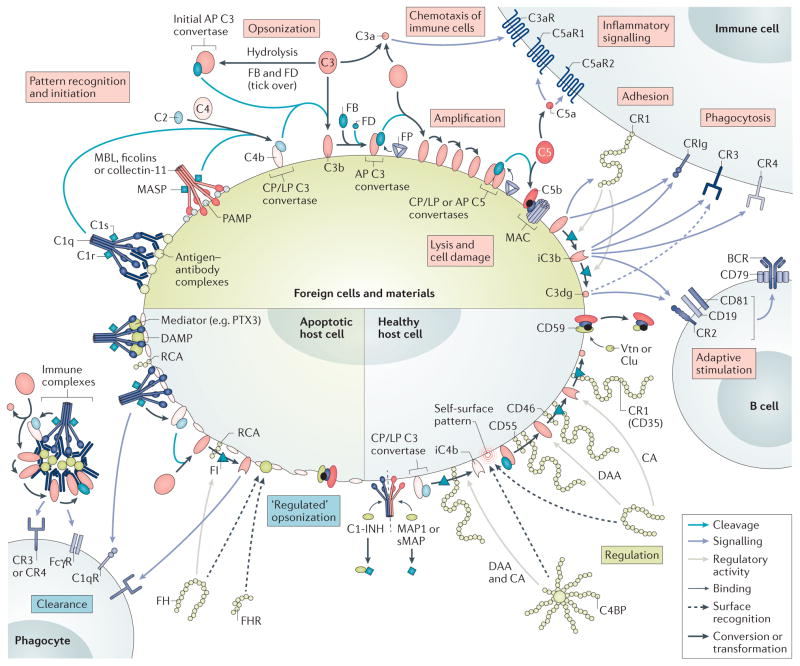
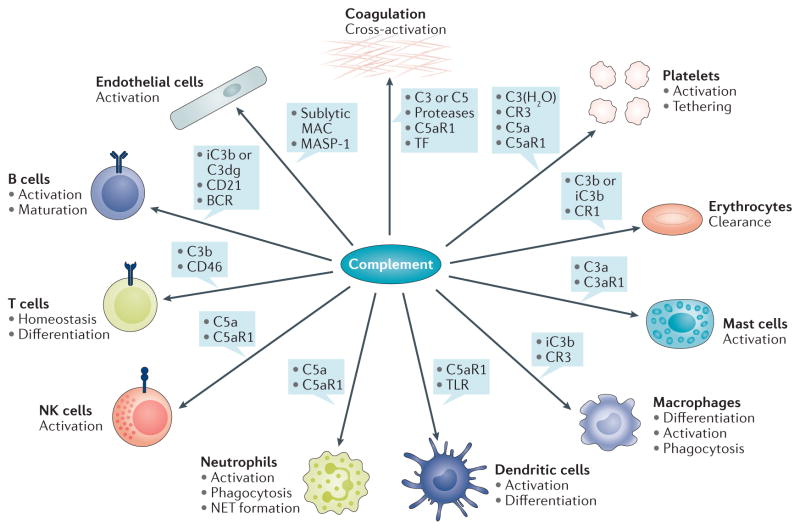
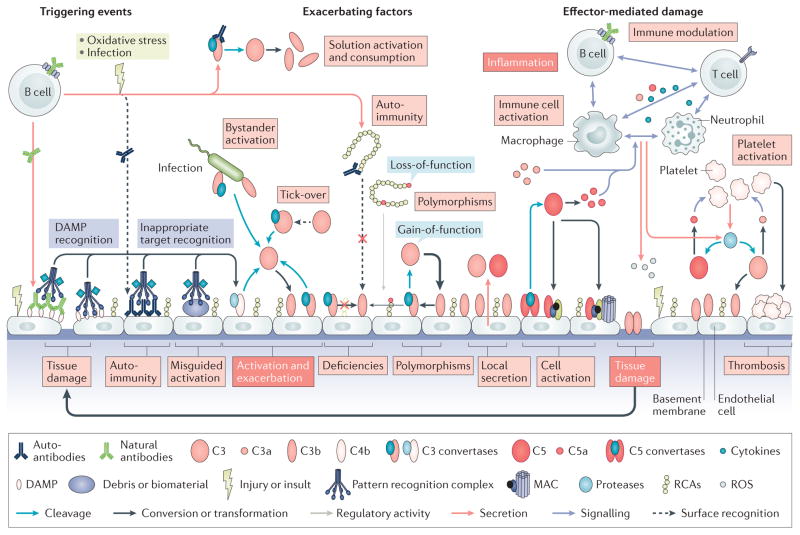
Although the complement system is primarily perceived as a host defence system, a more versatile, yet potentially more harmful side of this innate immune pathway as an inflammatory mediator also exists. The activities that define the ability of the complement system to control microbial threats and eliminate cellular debris - such as sensing molecular danger patterns, generating immediate effectors, and extensively coordinating with other defence pathways - can quickly turn complement from a defence system to an aggressor that drives immune and inflammatory diseases. These host-offensive actions become more pronounced with age and are exacerbated by a variety of genetic factors and autoimmune responses. Complement can also be activated inappropriately, for example in response to biomaterials or transplants. A wealth of research over the past two decades has led to an increasingly finely tuned understanding of complement activation, identified tipping points between physiological and pathological behaviour, and revealed avenues for therapeutic intervention. This Review summarizes our current view of the key activating, regulatory, and effector mechanisms of the complement system, highlighting important crosstalk connections, and, with an emphasis on kidney disease and transplantation, discusses the involvement of complement in clinical conditions and promising therapeutic approaches.
Conflict of interest statement
J.D.L and D.R. are inventors of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes. J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors. E.S.R. declares no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
