

TMEM5-associated dystroglycanopathy presenting with CMD and mild limb-girdle muscle involvement
- PMID: 27212206
- PMCID: PMC4925463
- DOI: 10.1016/j.nmd.2016.05.003
TMEM5-associated dystroglycanopathy presenting with CMD and mild limb-girdle muscle involvement
Abstract
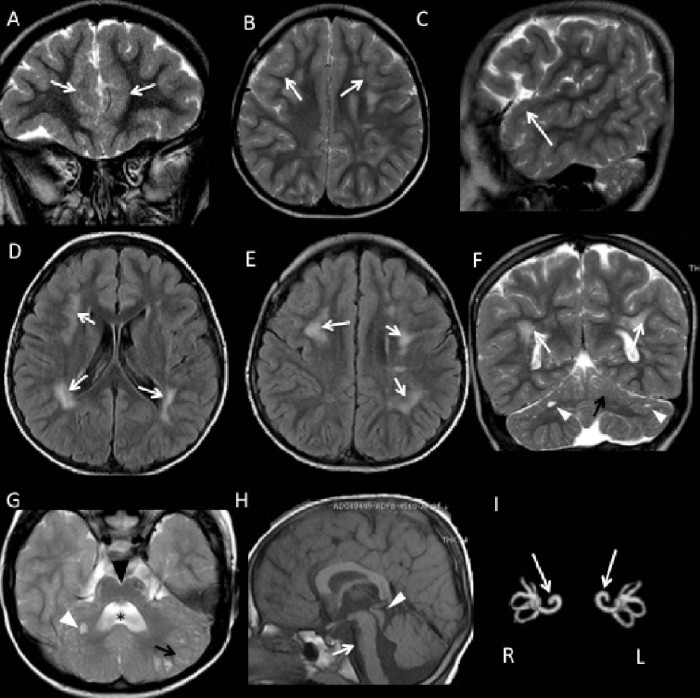
The dystroglycanopathies, which are caused by reduced glycosylation of alpha-dystroglycan, are a heterogeneous group of neurodegenerative disorders characterized by variable brain and skeletal muscle involvement. Recently, mutations in TMEM5 have been described in severe dystroglycanopathies. We present the clinical, molecular and neuroimaging features of an Italian boy who had delayed developmental milestones with mild limb-girdle muscle involvement, bilateral frontotemporal polymicrogyria, moderate intellectual disability, and no cerebellar involvement. He also presented a cochlear dysplasia and harbored a reported mutation (p.A47Rfs*42) in TMEM5, detected using targeted next-generation sequencing. The relatively milder muscular phenotype and associated structural brain abnormalities distinguish this case from previously reported patients with severe dystroglycanopathies and expand the spectrum of TMEM5-associated disorders.
Keywords: Cochlear dysplasia; Congenital muscular dystrophy; Limb-girdle muscle weakness; Polymicrogyria; TMEM5.
Copyright © 2016 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Muntoni F., Torelli S., Wells D.J., Brown S.C. Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies. Curr Opin Neurol. 2011;24:437–442. - PubMed
-
- Godfrey C., Clement E., Mein R. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
