

Spontaneous mutations and the origin and maintenance of quantitative genetic variation
- PMID: 27213517
- PMCID: PMC4929002
- DOI: 10.7554/eLife.14625
Spontaneous mutations and the origin and maintenance of quantitative genetic variation
Erratum in
-
Correction: Spontaneous mutations and the origin and maintenance of quantitative genetic variation.Elife. 2016 Oct 19;5:e22300. doi: 10.7554/eLife.22300. Elife. 2016. PMID: 27759564 Free PMC article. No abstract available.
Abstract
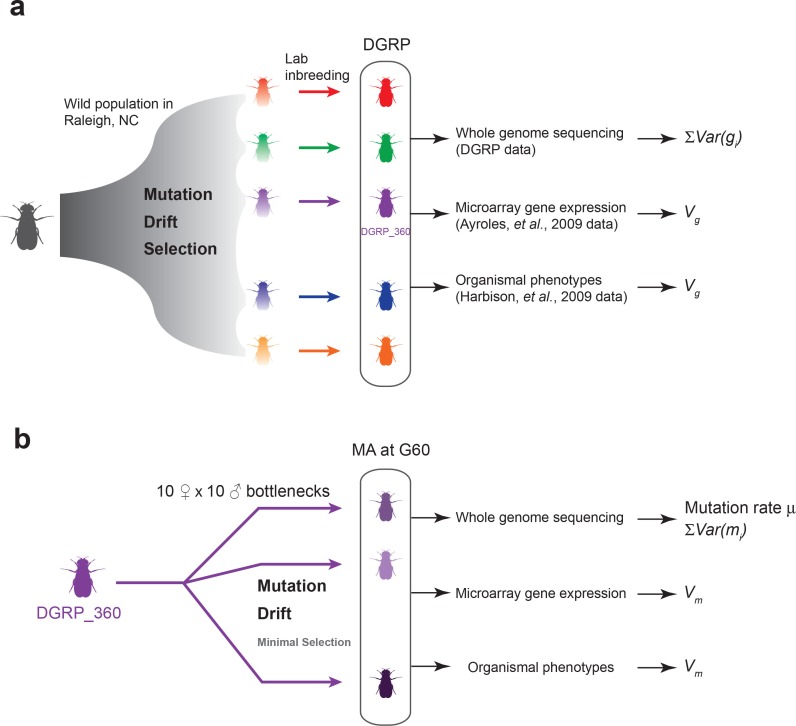
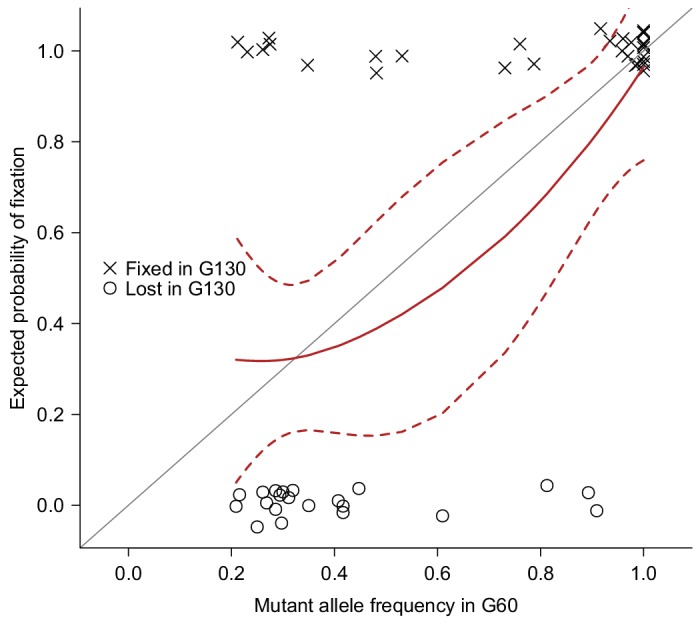
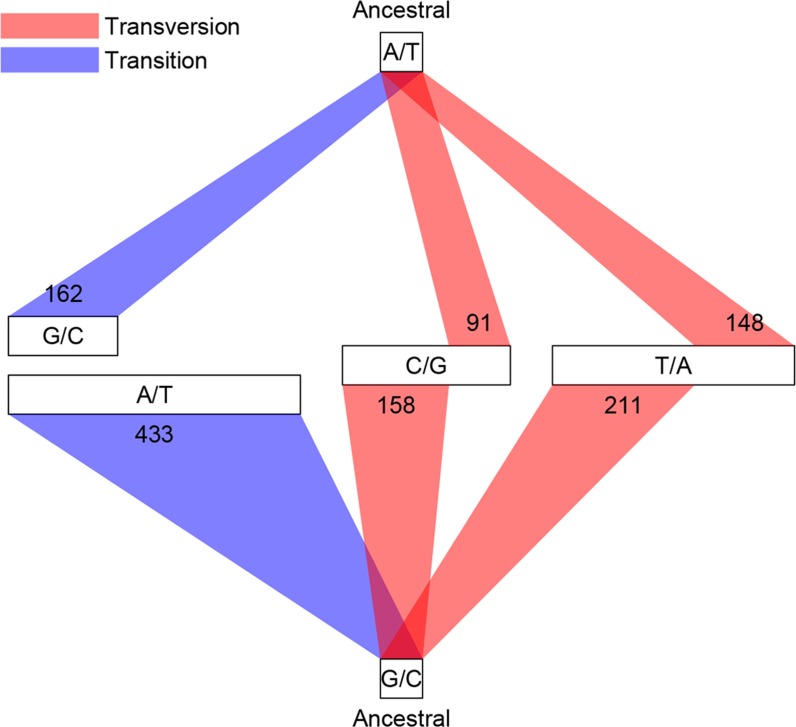
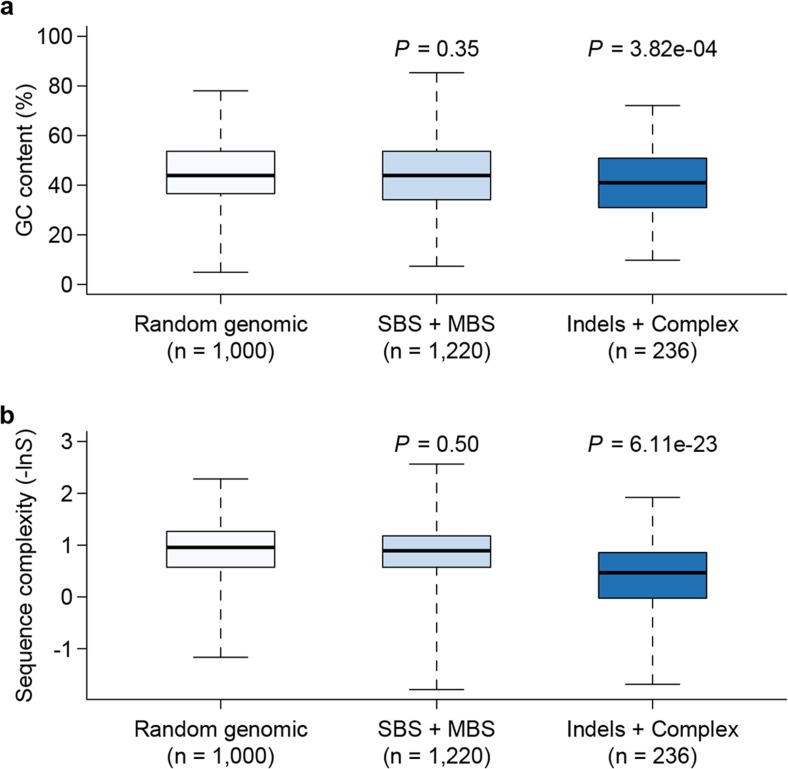
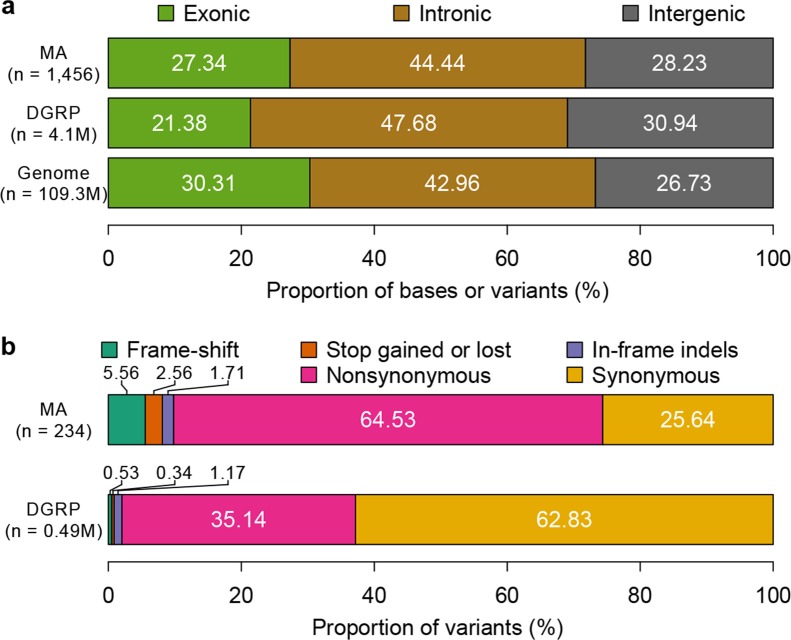
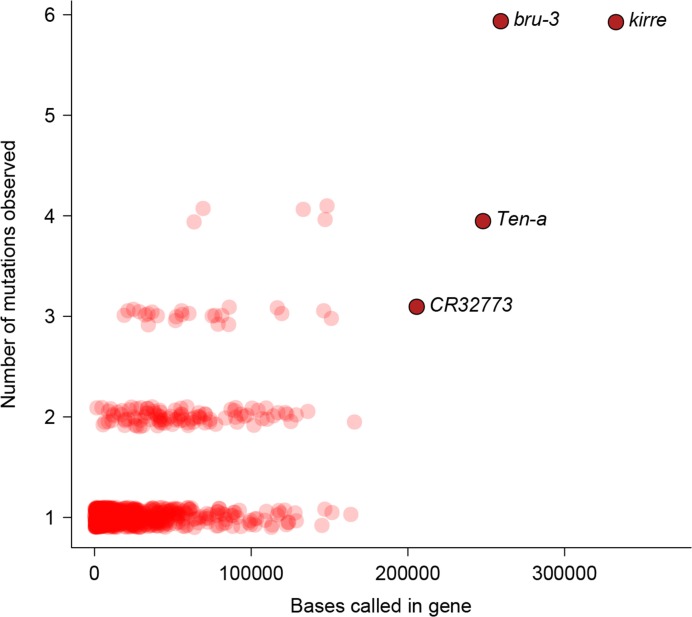
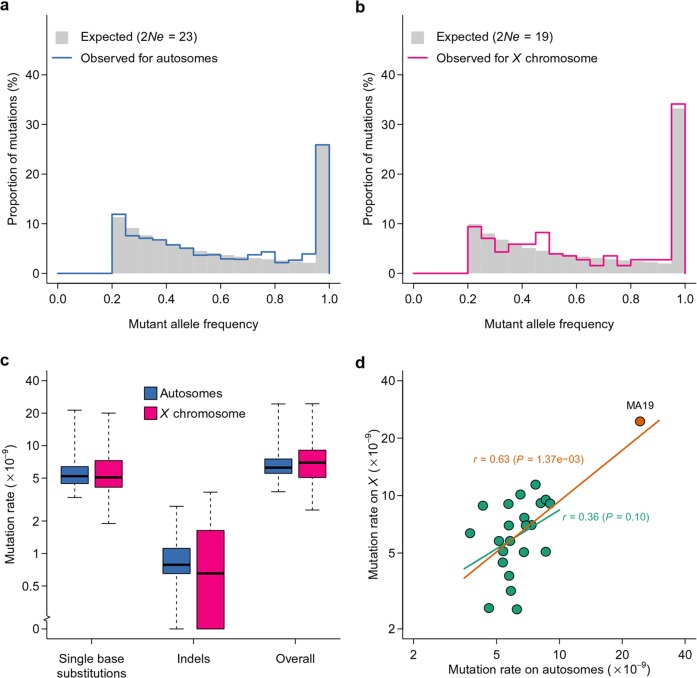
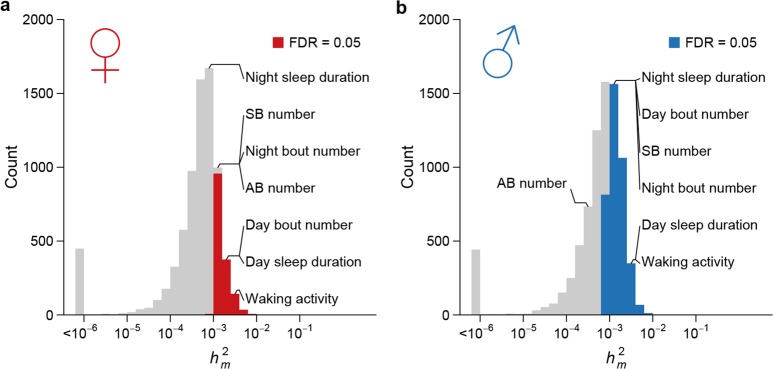
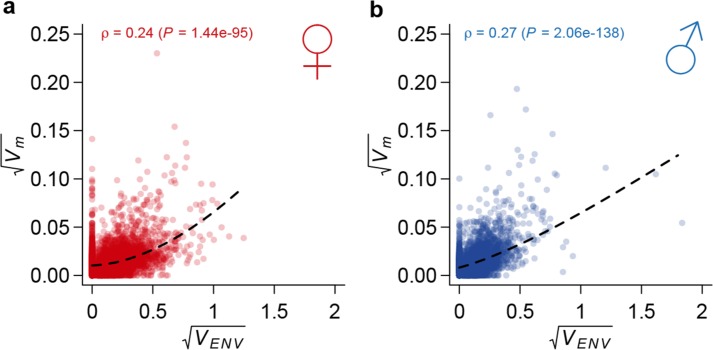
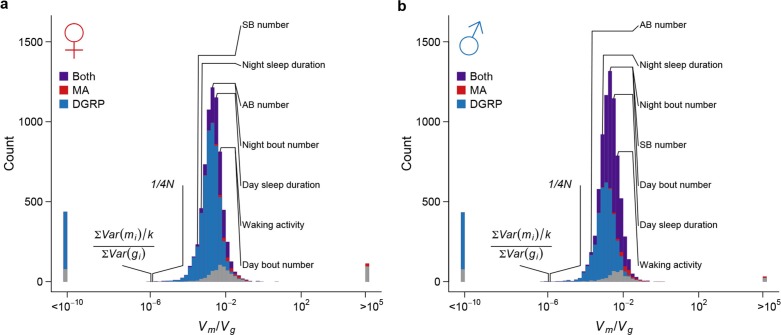
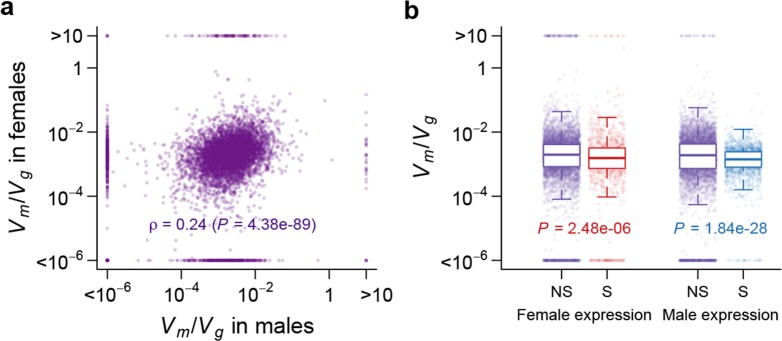
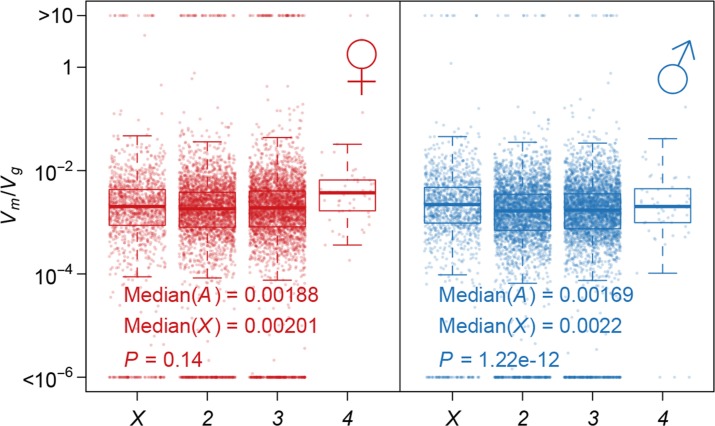
Mutation and natural selection shape the genetic variation in natural populations. Here, we directly estimated the spontaneous mutation rate by sequencing new Drosophila mutation accumulation lines maintained with minimal natural selection. We inferred strong stabilizing natural selection on quantitative traits because genetic variation among wild-derived inbred lines was much lower than predicted from a neutral model and the mutational effects were much larger than allelic effects of standing polymorphisms. Stabilizing selection could act directly on the traits, or indirectly from pleiotropic effects on fitness. However, our data are not consistent with simple models of mutation-stabilizing selection balance; therefore, further empirical work is needed to assess the balance of evolutionary forces responsible for quantitative genetic variation.
Keywords: D. melanogaster; evolutionary biology; gene expression; genome sequencing; genomics; mutation accumulation; stabilizing selection.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures
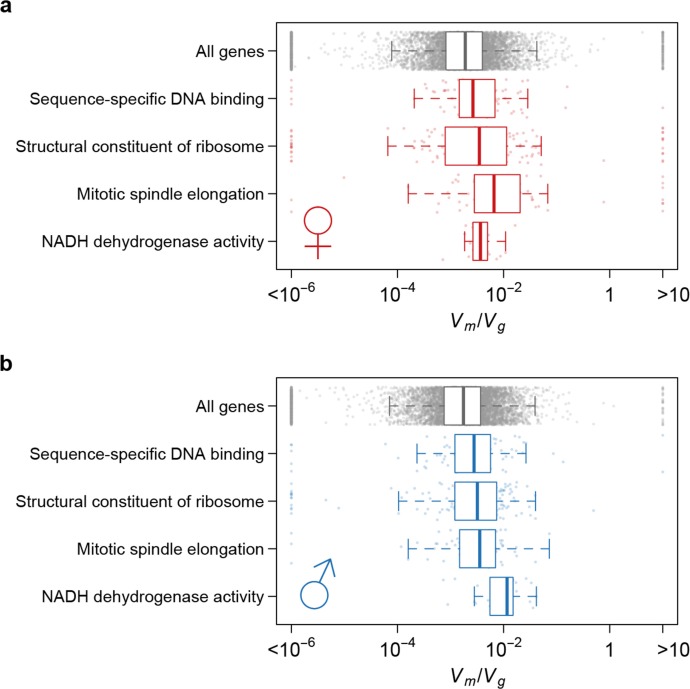
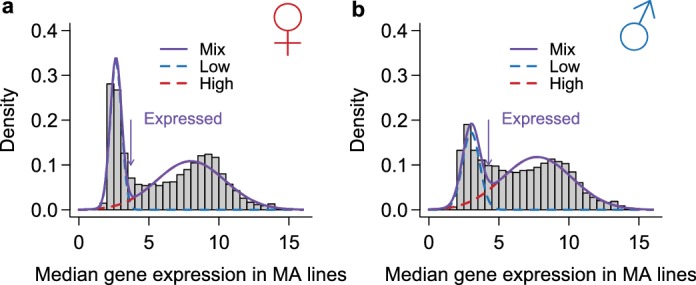
Similar articles
-
Mutations and quantitative genetic variation: lessons from Drosophila.Philos Trans R Soc Lond B Biol Sci. 2010 Apr 27;365(1544):1229-39. doi: 10.1098/rstb.2009.0315. Philos Trans R Soc Lond B Biol Sci. 2010. PMID: 20308098 Free PMC article. Review.
-
Causes of natural variation in fitness: evidence from studies of Drosophila populations.Proc Natl Acad Sci U S A. 2015 Feb 10;112(6):1662-9. doi: 10.1073/pnas.1423275112. Epub 2015 Jan 8. Proc Natl Acad Sci U S A. 2015. PMID: 25572964 Free PMC article. Review.
-
Polygenic mutation in Drosophila melanogaster: Mapping spontaneous mutations affecting sensory bristle number.Genetics. 2005 Aug;170(4):1723-35. doi: 10.1534/genetics.104.032581. Epub 2005 Jun 8. Genetics. 2005. PMID: 15944368 Free PMC article.
-
Influence of dominance, leptokurtosis and pleiotropy of deleterious mutations on quantitative genetic variation at mutation-selection balance.Genetics. 2004 Jan;166(1):597-610. doi: 10.1534/genetics.166.1.597. Genetics. 2004. PMID: 15020447 Free PMC article.
-
Pleiotropic mutations are subject to strong stabilizing selection.Genetics. 2014 Jul;197(3):1051-62. doi: 10.1534/genetics.114.165720. Epub 2014 May 2. Genetics. 2014. PMID: 24793289 Free PMC article.
Cited by
-
Negative frequency-dependent selection maintains coexisting genotypes during fluctuating selection.Mol Ecol. 2020 Jan;29(1):138-148. doi: 10.1111/mec.15307. Epub 2019 Dec 5. Mol Ecol. 2020. PMID: 31725941 Free PMC article.
-
Old Trade, New Tricks: Insights into the Spontaneous Mutation Process from the Partnering of Classical Mutation Accumulation Experiments with High-Throughput Genomic Approaches.Genome Biol Evol. 2019 Jan 1;11(1):136-165. doi: 10.1093/gbe/evy252. Genome Biol Evol. 2019. PMID: 30476040 Free PMC article. Review.
-
Effect of genetic architecture on the prediction accuracy of quantitative traits in samples of unrelated individuals.Heredity (Edinb). 2018 Jun;120(6):500-514. doi: 10.1038/s41437-017-0043-0. Epub 2018 Feb 10. Heredity (Edinb). 2018. PMID: 29426878 Free PMC article.
-
Genomes from historical Drosophila melanogaster specimens illuminate adaptive and demographic changes across more than 200 years of evolution.PLoS Biol. 2023 Oct 12;21(10):e3002333. doi: 10.1371/journal.pbio.3002333. eCollection 2023 Oct. PLoS Biol. 2023. PMID: 37824452 Free PMC article.
-
Spontaneous single-nucleotide substitutions and microsatellite mutations have distinct distributions of fitness effects.PLoS Biol. 2024 Jul 1;22(7):e3002698. doi: 10.1371/journal.pbio.3002698. eCollection 2024 Jul. PLoS Biol. 2024. PMID: 38950062 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
