

Why factor XI deficiency is a clinical concern
- PMID: 27216469
- PMCID: PMC4950943
- DOI: 10.1080/17474086.2016.1191944
Why factor XI deficiency is a clinical concern
Abstract
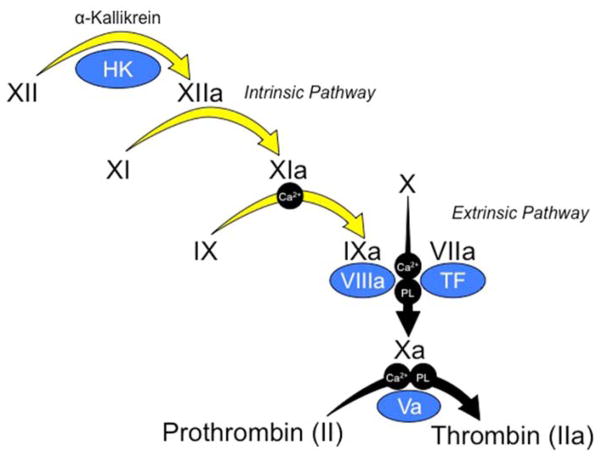
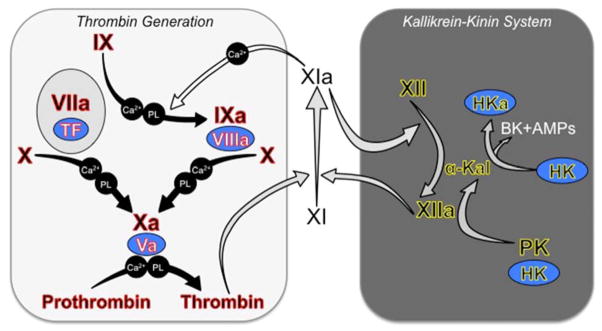
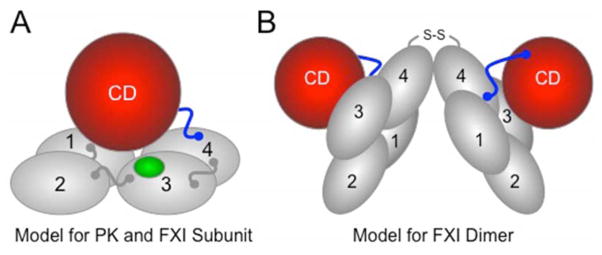
Introduction: Inherited fXI deficiency has been an enigma since its discovery in 1953. The variable and relatively mild symptoms in patients with even the most severe form of the disorder seem out of step with the marked abnormalities in standard clotting assays. Indeed, the contribution of factor XI to hemostasis in an individual is not adequately assessed by techniques available in modern clinical laboratories.
Areas covered: We discuss clinical studies, genetic/genomic analyses, and advances in laboratory medicine that are reshaping our views on the role of factor XI in pathologic coagulation. We review how the disorder associated with factor XI deficiency has contributed to changes in blood coagulation models, and discuss the complex genetics of the deficiency state and its relationship to bleeding. Finally, we cover new laboratory approaches that may distinguish deficient patients who are prone to bleeding from those without such predisposition. Expert commentary: Advances in understanding the biology of factor XI have led to modifications in treatment of factor XI-deficient patients. Factor replacement is used more judiciously, and alternative approaches are gaining favor. In the future, better laboratory tests may allow us to target therapy to those patients who would benefit most.
Keywords: Factor XI; contact activation; factor XI deficiency; factor XII; prekallikrein.
Figures
References
-
- Rosenthal RL, Dreskin OH, Rosenthal N. New hemophilia-like disease caused by deficiency of a third plasma thromboplastin factor. Proc Soc Exp Biol Med. 1953;82:171–4. - PubMed
-
- Rosenthal RL, Dreskin OH, Rosenthal N. Plasma thromboplastin antecedent (PTA) deficiency; clinical, coagulation, therapeutic and hereditary aspects of a new hemophilia-like disease. Blood. 1955;10:120–31. - PubMed
-
- Duga S, Salomon O. Congenital factor XI deficiency: an update. Sem Thromb Hemost. 2013;39:621–31. - PubMed
-
- Santoro C, Di Mauro R, Baldacci E, et al. Bleeding phenotype and correlation with factor XI (FXI) activity in congenital FXI deficiency: results of a retrospective study from a single centre. Haemophilia. 2015;21:496–501. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous