

The FOXG1/FOXO/SMAD network balances proliferation and differentiation of cortical progenitors and activates Kcnh3 expression in mature neurons
- PMID: 27224923
- PMCID: PMC5122323
- DOI: 10.18632/oncotarget.9545
The FOXG1/FOXO/SMAD network balances proliferation and differentiation of cortical progenitors and activates Kcnh3 expression in mature neurons
Abstract
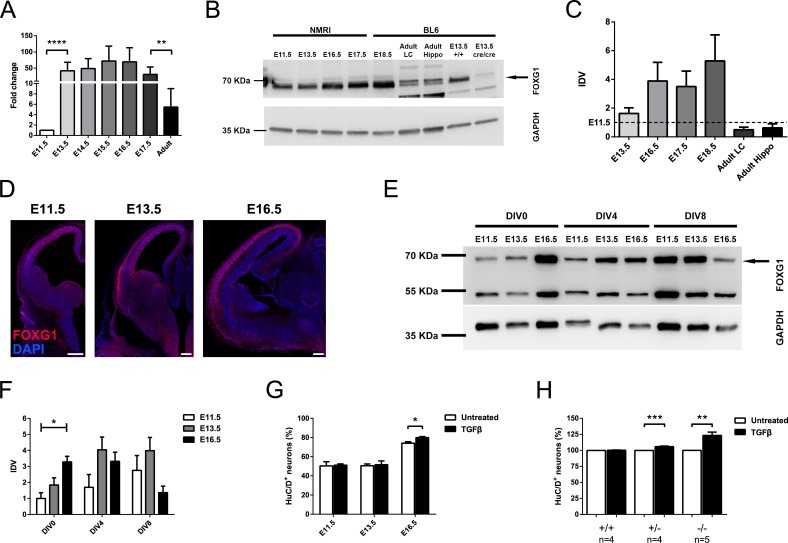
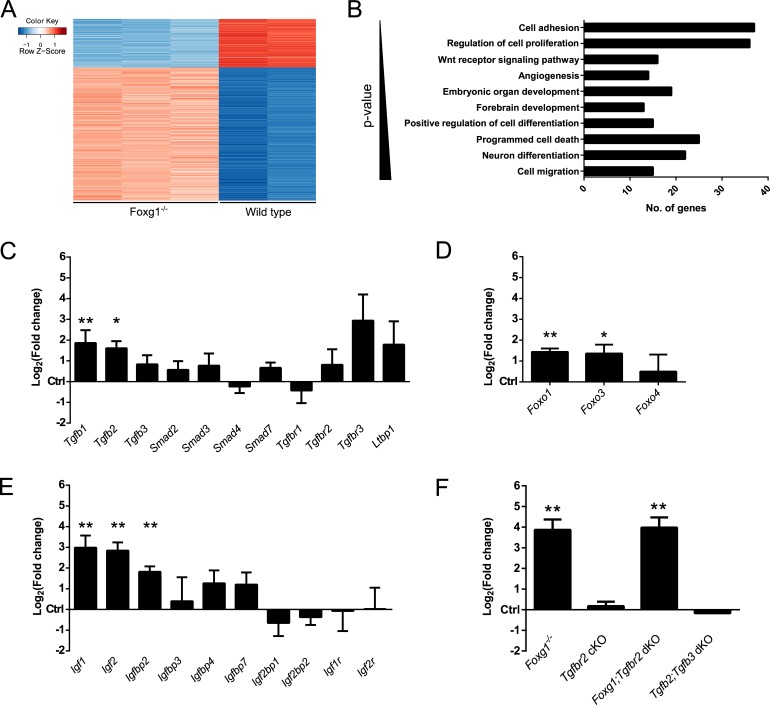
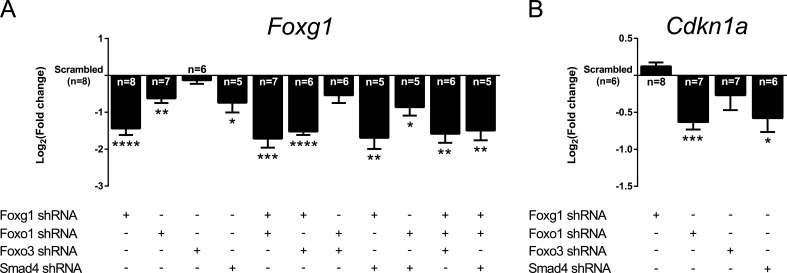
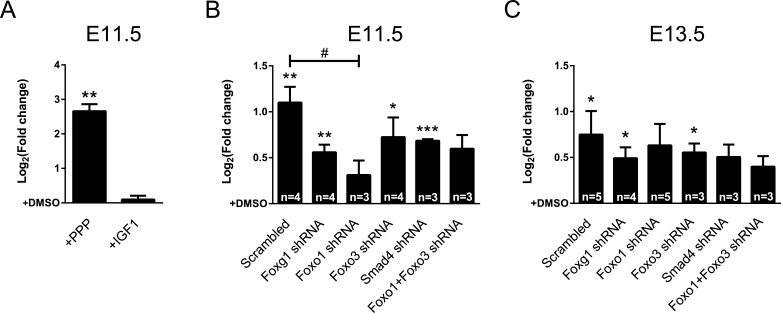
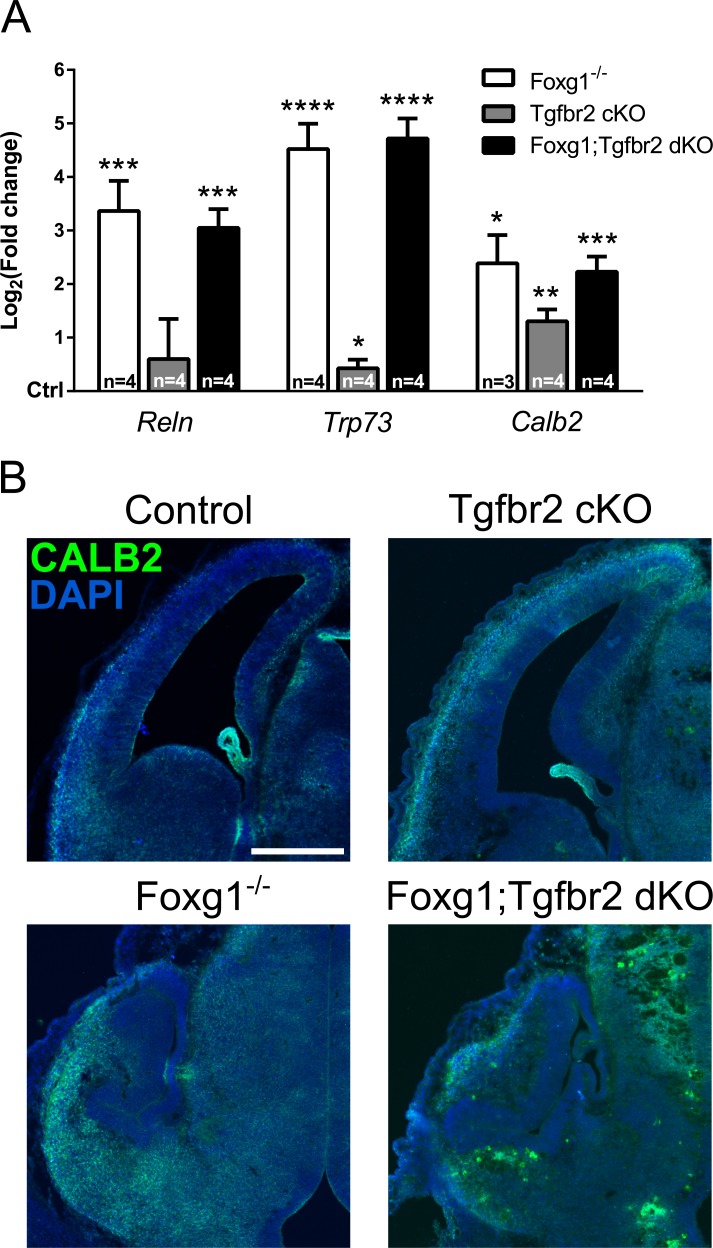
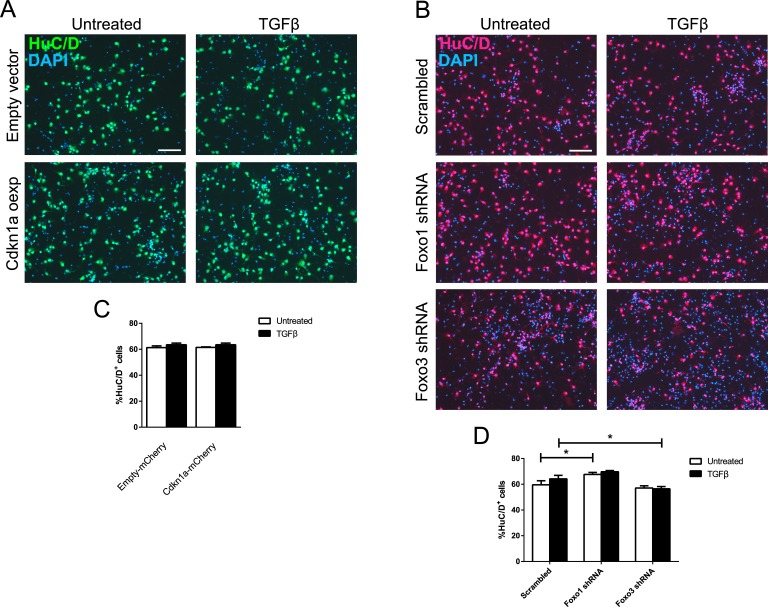
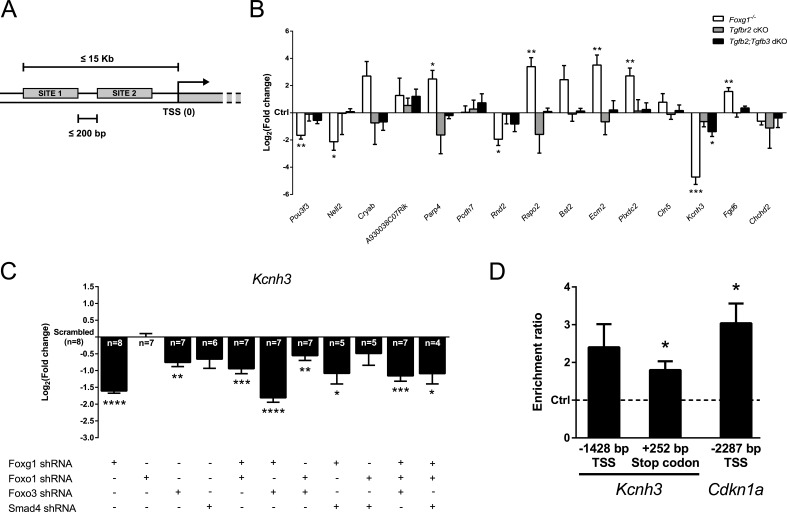
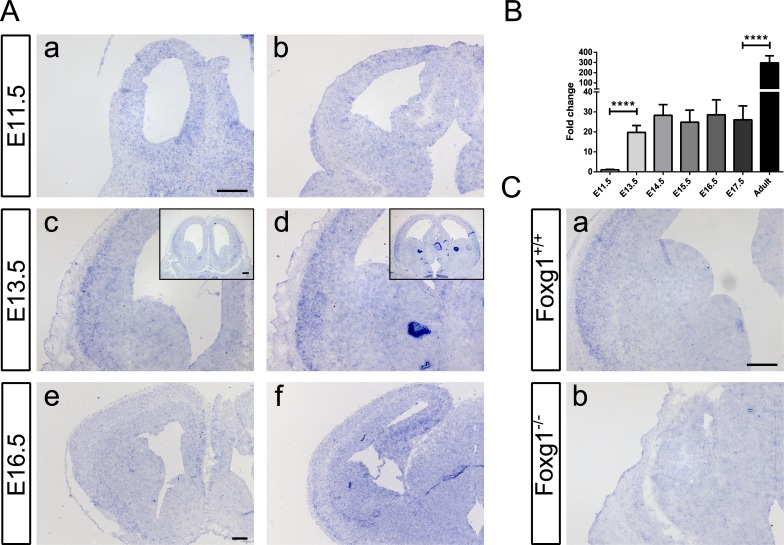
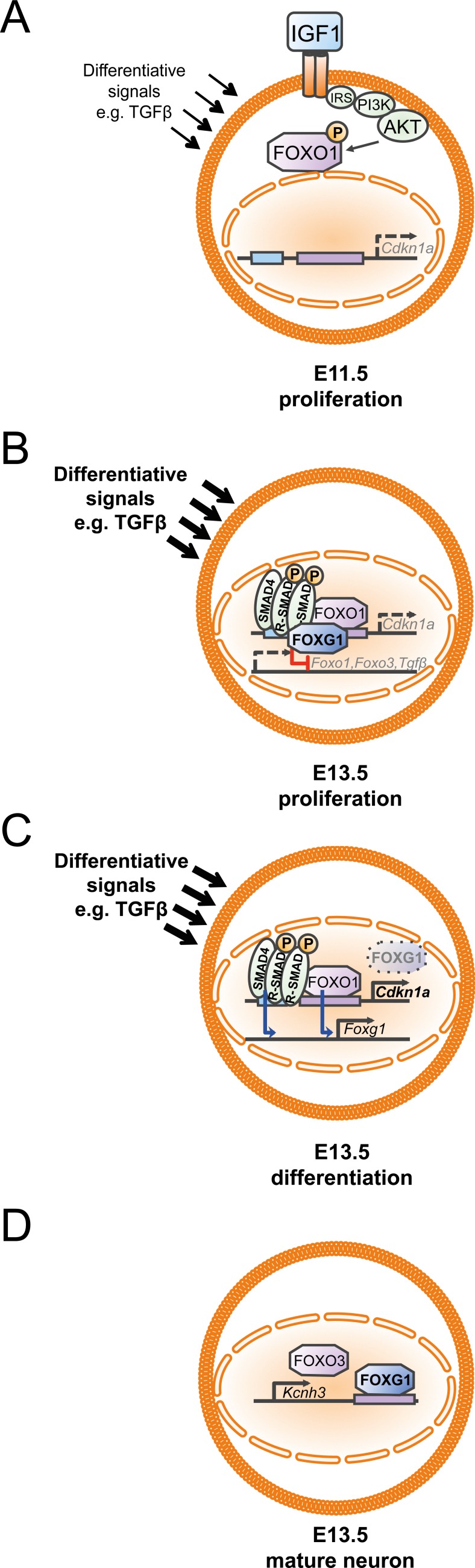
Transforming growth factor β (TGFβ)-mediated anti-proliferative and differentiating effects promote neuronal differentiation during embryonic central nervous system development. TGFβ downstream signals, composed of activated SMAD2/3, SMAD4 and a FOXO family member, promote the expression of cyclin-dependent kinase inhibitor Cdkn1a. In early CNS development, IGF1/PI3K signaling and the transcription factor FOXG1 inhibit FOXO- and TGFβ-mediated Cdkn1a transcription. FOXG1 prevents cell cycle exit by binding to the SMAD/FOXO-protein complex. In this study we provide further details on the FOXG1/FOXO/SMAD transcription factor network. We identified ligands of the TGFβ- and IGF-family, Foxo1, Foxo3 and Kcnh3 as novel FOXG1-target genes during telencephalic development and showed that FOXG1 interferes with Foxo1 and Tgfβ transcription. Our data specify that FOXO1 activates Cdkn1a transcription. This process is under control of the IGF1-pathway, as Cdkn1a transcription increases when IGF1-signaling is pharmacologically inhibited. However, overexpression of CDKN1A and knockdown of Foxo1 and Foxo3 is not sufficient for neuronal differentiation, which is probably instructed by TGFβ-signaling. In mature neurons, FOXG1 activates transcription of the seizure-related Kcnh3, which might be a FOXG1-target gene involved in the FOXG1 syndrome pathology.
Keywords: Gerotarget; TGFβ; atypical Rett syndrome; cerebral cortex; neurogenesis; transcriptional control.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Figures
References
-
- Vogel T, Ahrens S, Büttner N, Krieglstein K. Transforming growth factor beta promotes neuronal cell fate of mouse cortical and hippocampal progenitors in vitro and in vivo: identification of Nedd9 as an essential signaling component. Cereb Cortex. 2010;20:661–71. doi: 10.1093/cercor/bhp134. - DOI - PMC - PubMed
-
- Wahane SD, Hellbach N, Prentzell MT, Weise SC, Vezzali R, Kreutz C, Timmer J, Krieglstein K, Thedieck K, Vogel T. PI3K-p110-alpha-subtype signalling mediates survival, proliferation and neurogenesis of cortical progenitor cells via activation of mTORC2. J Neurochem. 2014;130:255–67. doi: 10.1111/jnc.12718. - DOI - PubMed
-
- Dünker N, Krieglstein K. Targeted mutations of transforming growth factor-beta genes reveal important roles in mouse development and adult homeostasis. Eur J Biochem. 2000;267:6982–8. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
