

Chromatin dynamics during DNA replication
- PMID: 27225843
- PMCID: PMC5052047
- DOI: 10.1101/gr.201244.115
Chromatin dynamics during DNA replication
Abstract
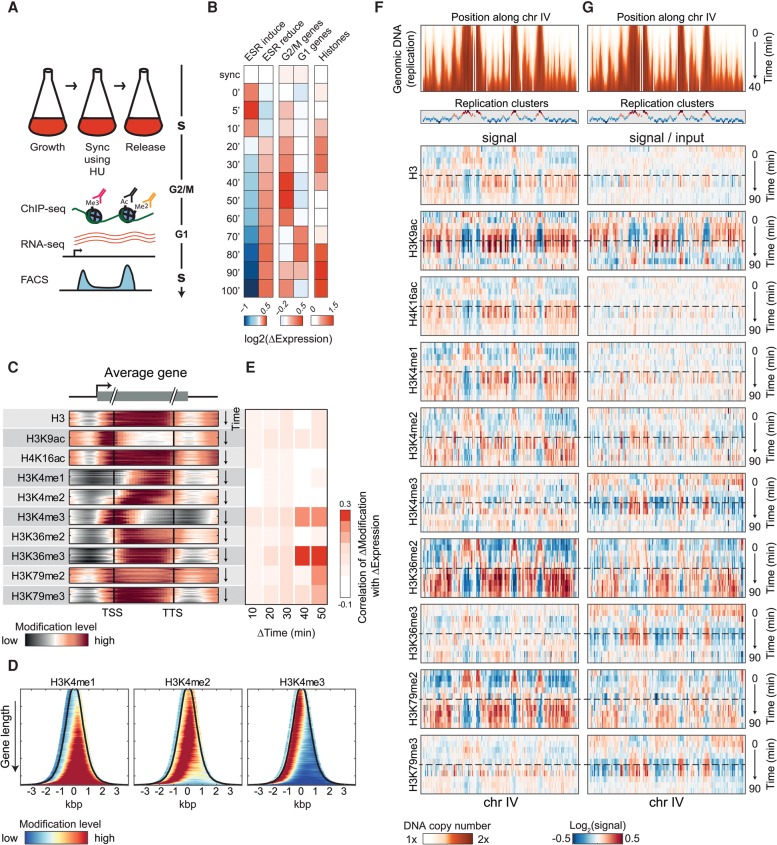
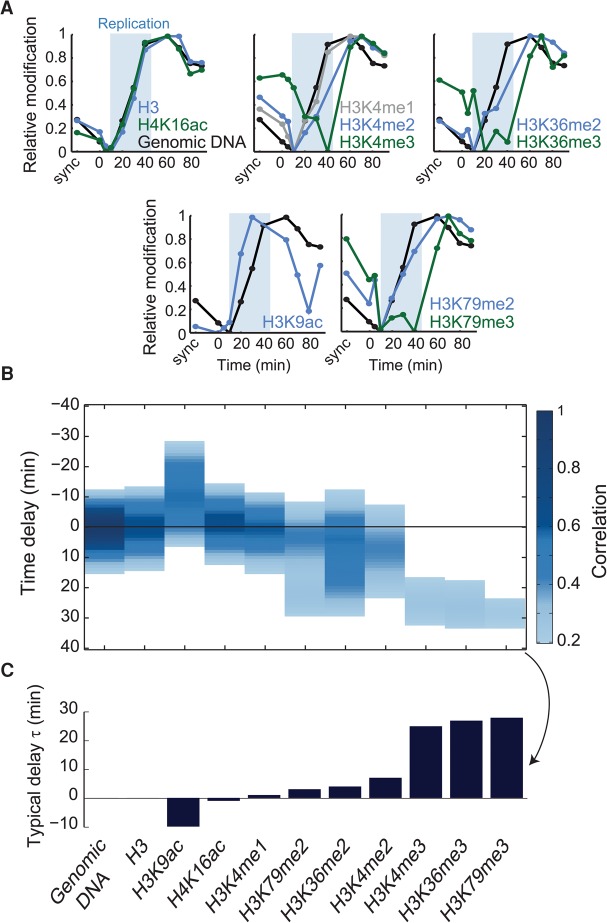
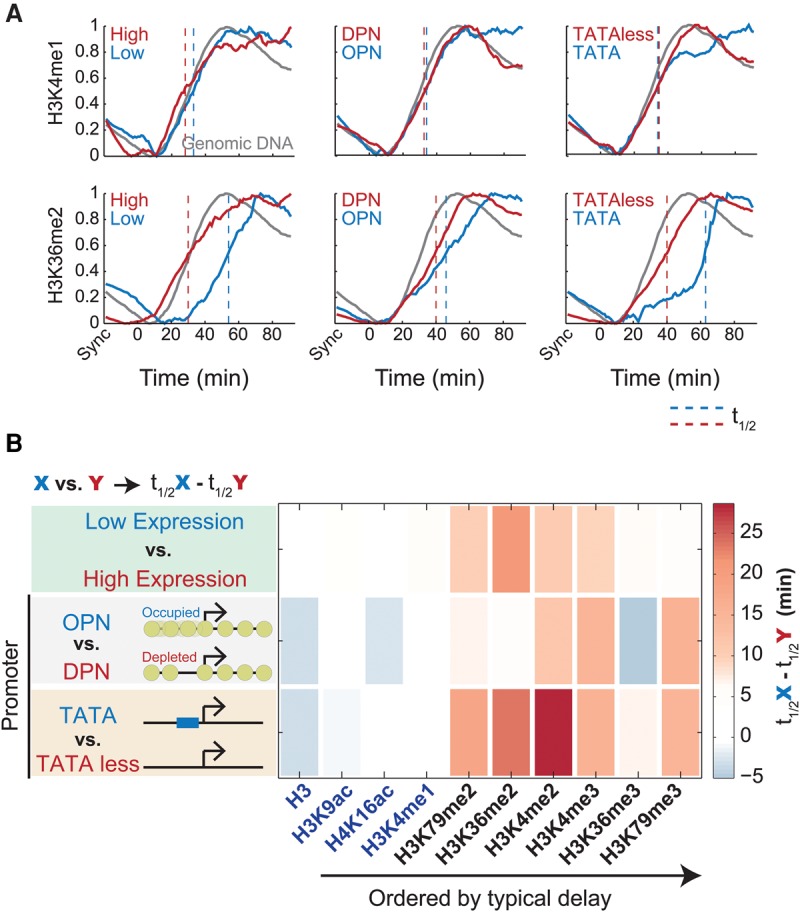
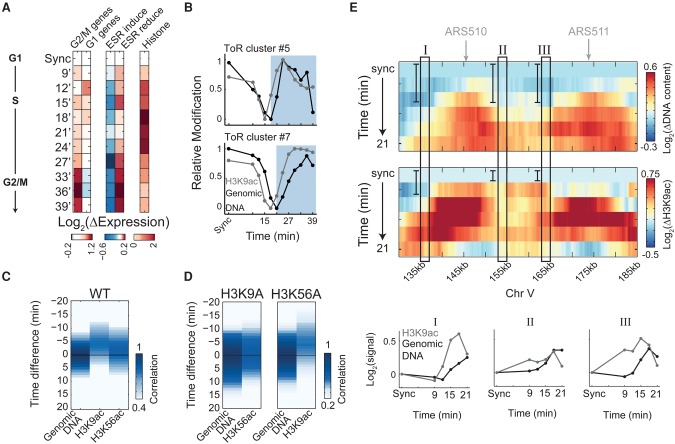
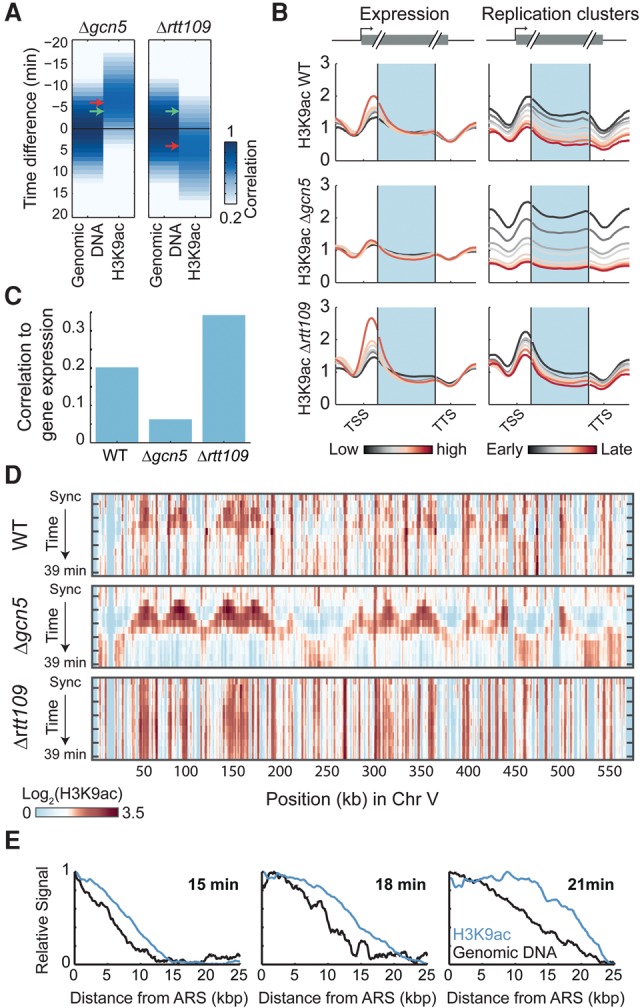
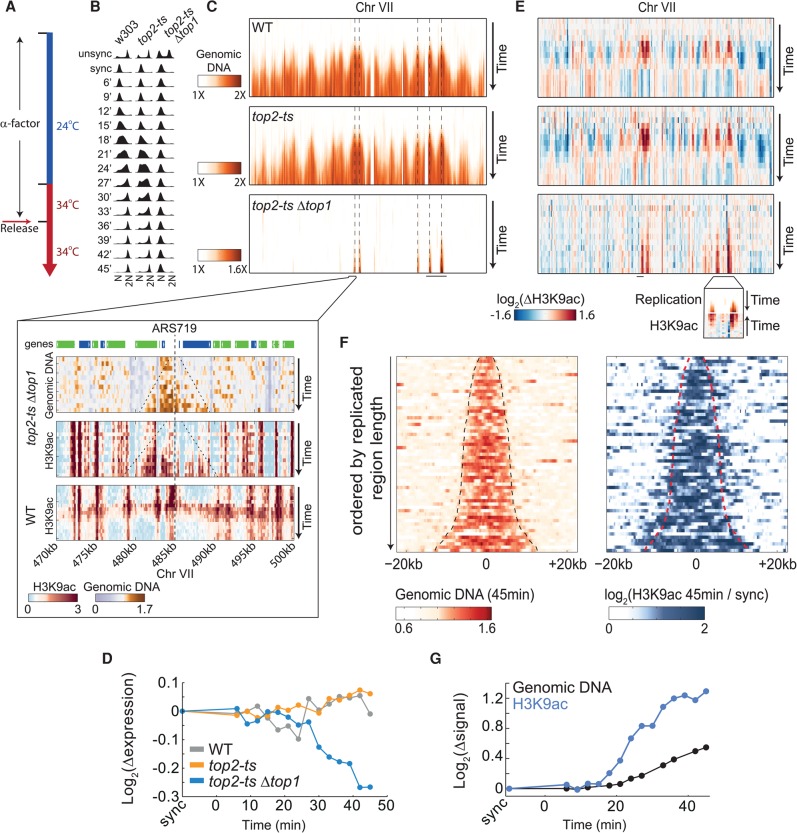
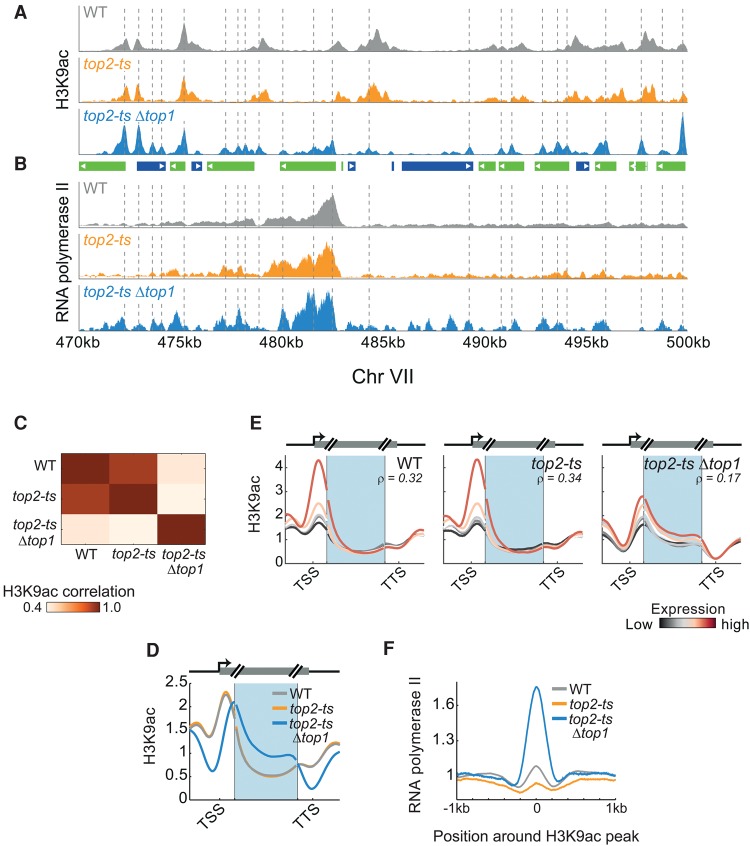
Chromatin is composed of DNA and histones, which provide a unified platform for regulating DNA-related processes, mostly through their post-translational modification. During DNA replication, histone arrangement is perturbed, first to allow progression of DNA polymerase and then during repackaging of the replicated DNA. To study how DNA replication influences the pattern of histone modification, we followed the cell-cycle dynamics of 10 histone marks in budding yeast. We find that histones deposited on newly replicated DNA are modified at different rates: While some marks appear immediately upon replication (e.g., H4K16ac, H3K4me1), others increase with transcription-dependent delays (e.g., H3K4me3, H3K36me3). Notably, H3K9ac was deposited as a wave preceding the replication fork by ∼5-6 kb. This replication-guided H3K9ac was fully dependent on the acetyltransferase Rtt109, while expression-guided H3K9ac was deposited by Gcn5. Further, topoisomerase depletion intensified H3K9ac in front of the replication fork and in sites where RNA polymerase II was trapped, suggesting supercoiling stresses trigger H3K9 acetylation. Our results assign complementary roles for DNA replication and gene expression in defining the pattern of histone modification.
© 2016 Bar-Ziv et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Annunziato AT. 2005. Split decision: What happens to nucleosomes during DNA replication? J Biol Chem 280: 12065–12068. - PubMed
-
- Baxter J, Diffley JFX. 2008. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell 30: 790–802. - PubMed
-
- Benson LJ, Gu Y, Yakovleva T, Tong K, Barrows C, Strack CL, Cook RG, Mizzen CA, Annunziato AT. 2006. Modifications of H3 and H4 during chromatin replication, nucleosome assembly, and histone exchange. J Biol Chem 281: 9287–9296. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases