

Whole-organism lineage tracing by combinatorial and cumulative genome editing
- PMID: 27229144
- PMCID: PMC4967023
- DOI: 10.1126/science.aaf7907
Whole-organism lineage tracing by combinatorial and cumulative genome editing
Abstract
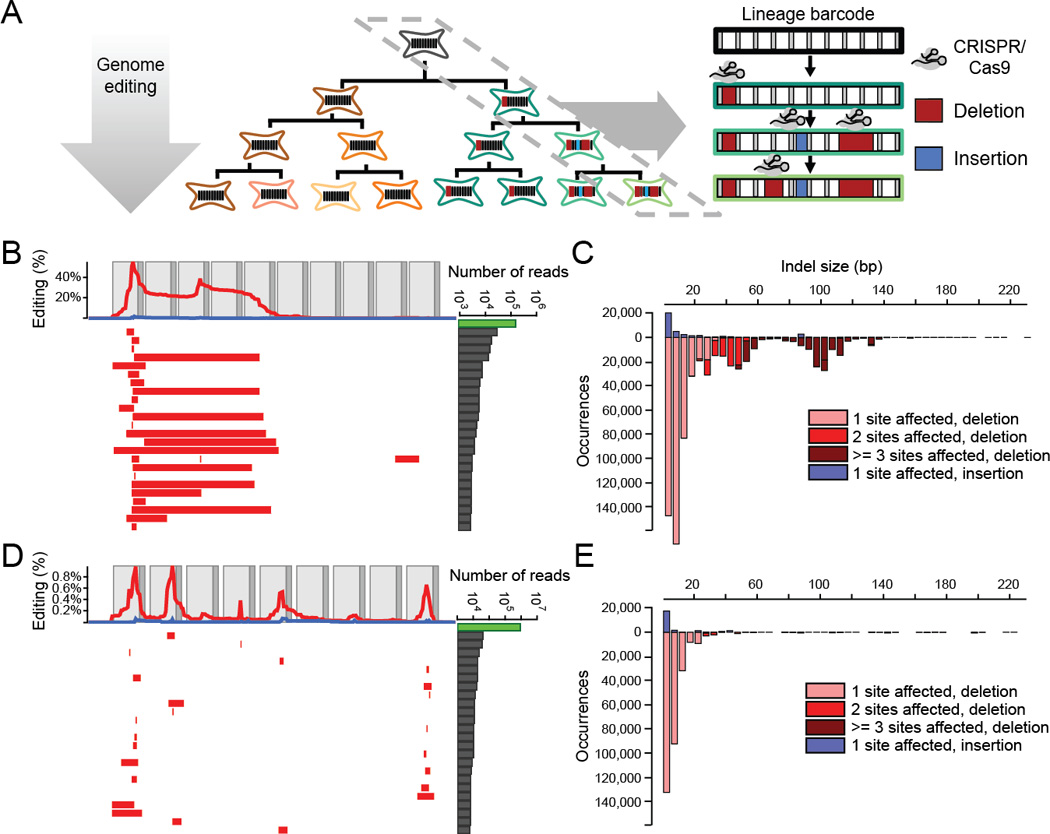
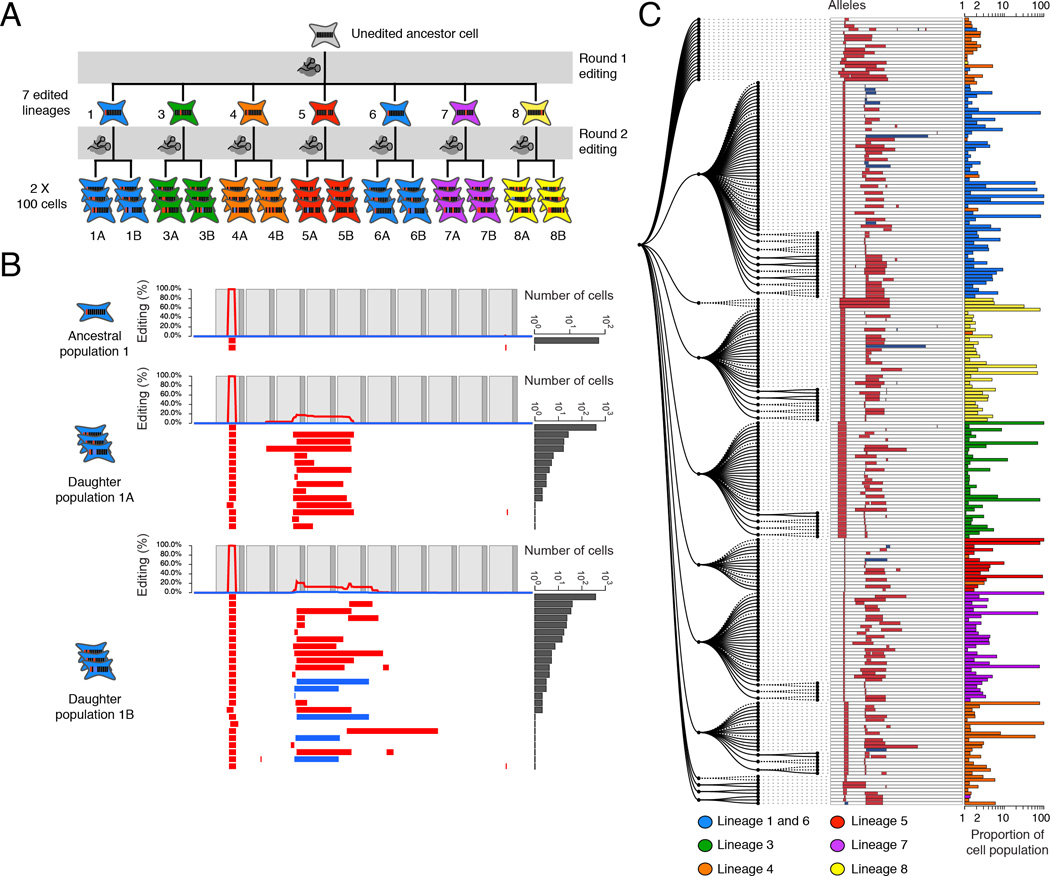
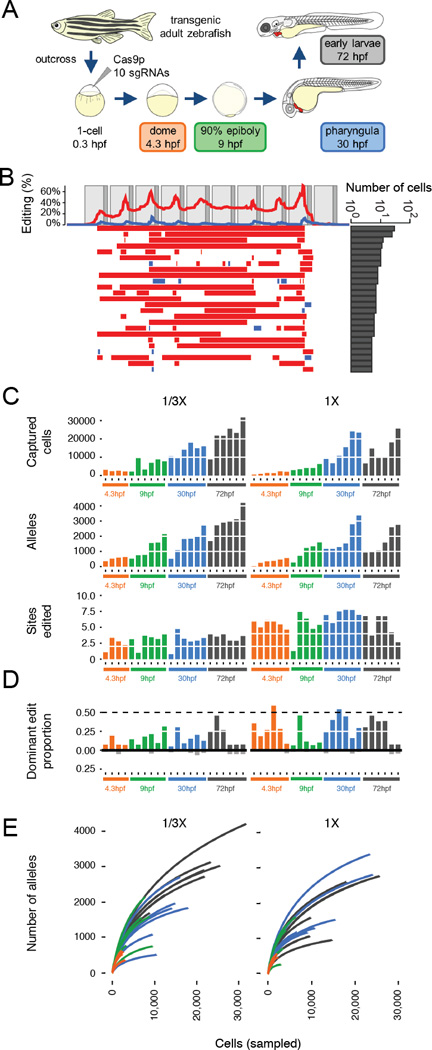
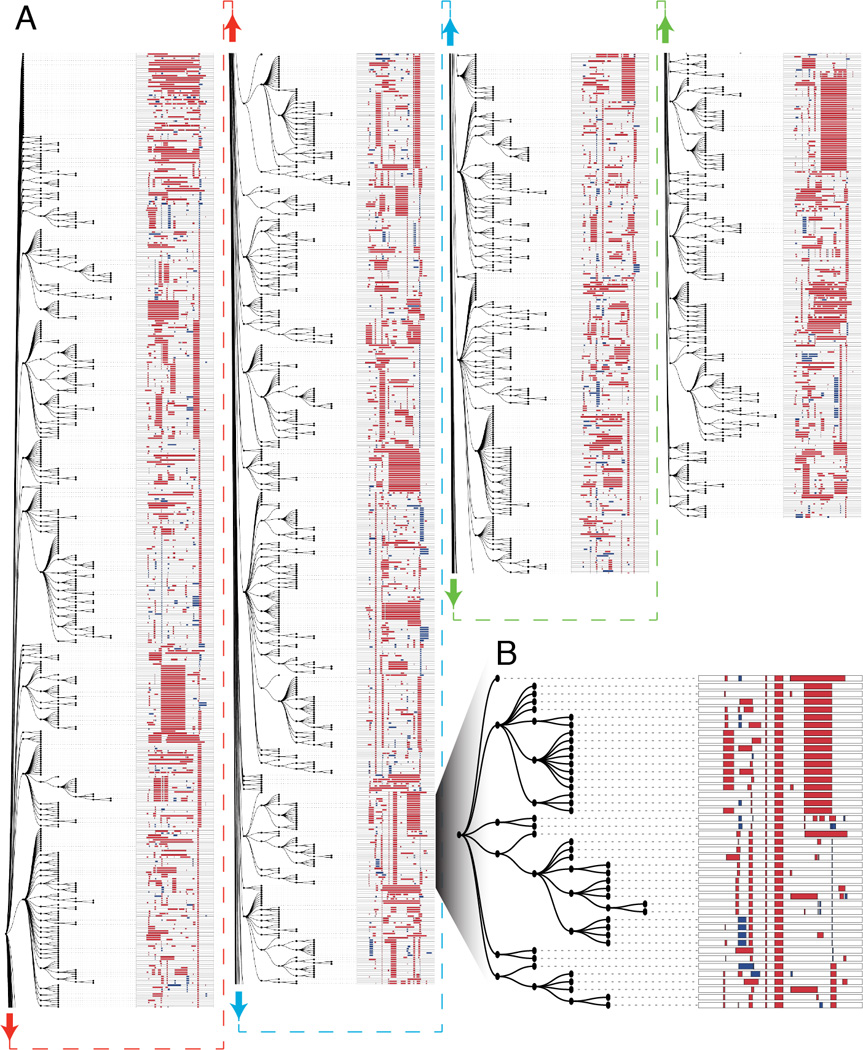
Multicellular systems develop from single cells through distinct lineages. However, current lineage-tracing approaches scale poorly to whole, complex organisms. Here, we use genome editing to progressively introduce and accumulate diverse mutations in a DNA barcode over multiple rounds of cell division. The barcode, an array of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 target sites, marks cells and enables the elucidation of lineage relationships via the patterns of mutations shared between cells. In cell culture and zebrafish, we show that rates and patterns of editing are tunable and that thousands of lineage-informative barcode alleles can be generated. By sampling hundreds of thousands of cells from individual zebrafish, we find that most cells in adult organs derive from relatively few embryonic progenitors. In future analyses, genome editing of synthetic target arrays for lineage tracing (GESTALT) can be used to generate large-scale maps of cell lineage in multicellular systems for normal development and disease.
Copyright © 2016, American Association for the Advancement of Science.
Figures
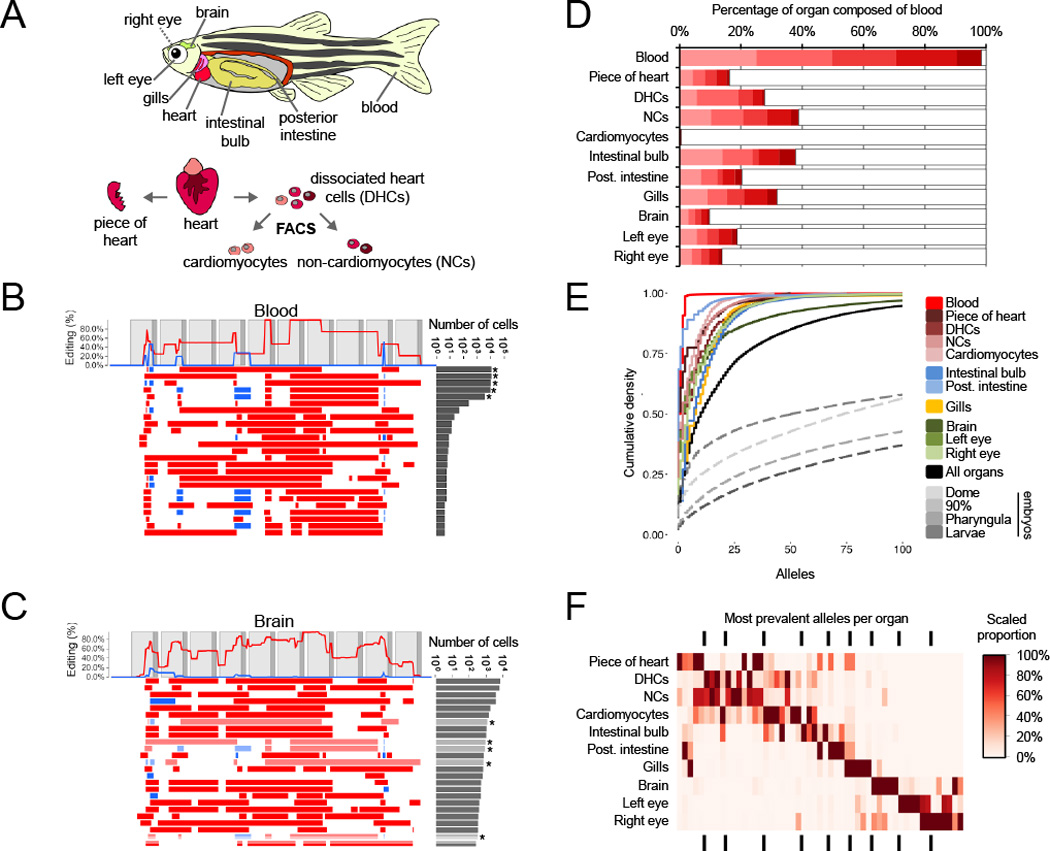
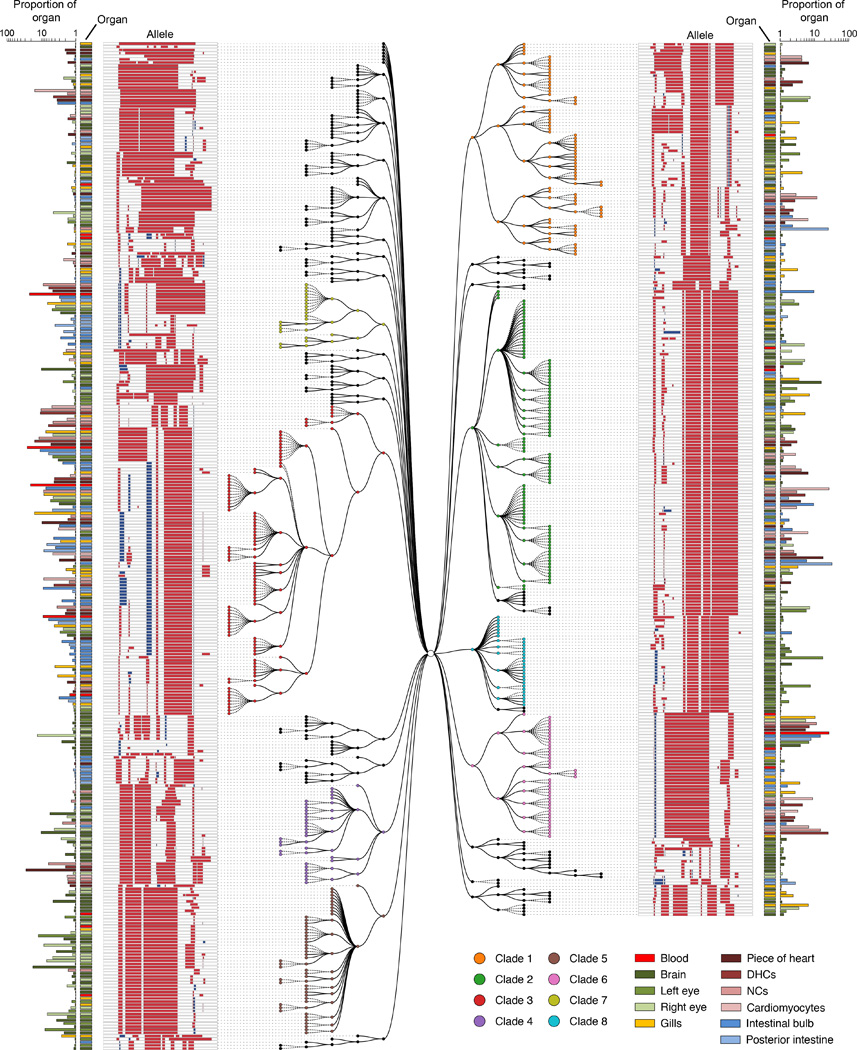
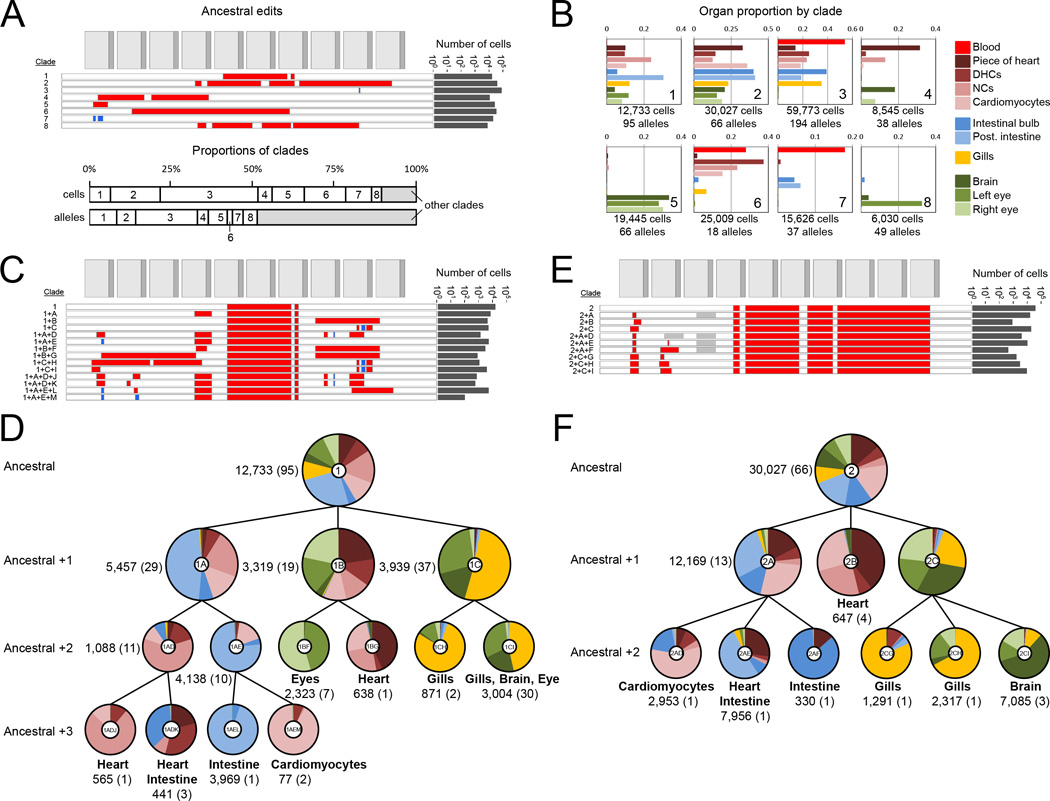
Comment in
-
Tracing cell lineages with mutable barcodes.Nat Biotechnol. 2016 Jul 12;34(7):725. doi: 10.1038/nbt.3634. Nat Biotechnol. 2016. PMID: 27404885 No abstract available.
References
-
- Stent GS. Developmental cell lineage. Int. J. Dev. Biol. 1998;42:237–241. - PubMed
-
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology. 1983;100:64–119. - PubMed
-
- Kretzschmar K, Watt FM. Lineage Tracing. Cell. 2012;148:33–45. - PubMed
-
- Kimmel CB, Law RD. Cell lineage of zebrafish blastomeres. III. Clonal analyses of the blastula and gastrula stages. Developmental Biology. 1985;108:94–101. - PubMed
-
- Keller RE. Vital dye mapping of the gastrula and neurula of Xenopus laevis. I. Prospective areas and morphogenetic movements of the superficial layer. Developmental Biology. 1975;42:222–241. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- MH105960/MH/NIMH NIH HHS/United States
- T32HL007312/HL/NHLBI NIH HHS/United States
- GM056211/GM/NIGMS NIH HHS/United States
- T32 HL007312/HL/NHLBI NIH HHS/United States
- R37 GM056211/GM/NIGMS NIH HHS/United States
- DP1HG007811/DP/NCCDPHP CDC HHS/United States
- U01 MH105960/MH/NIMH NIH HHS/United States
- R01 HD085905/HD/NICHD NIH HHS/United States
- R01 GM056211/GM/NIGMS NIH HHS/United States
- Howard Hughes Medical Institute/United States
- HD085905/HD/NICHD NIH HHS/United States
- T32 GM007266/GM/NIGMS NIH HHS/United States
- DP1 HG007811/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
