

Early pulmonary disease manifestations in cystic fibrosis mice
- PMID: 27231029
- PMCID: PMC5121081
- DOI: 10.1016/j.jcf.2016.05.002
Early pulmonary disease manifestations in cystic fibrosis mice
Abstract
Background: Altered pulmonary function is present early in the course of cystic fibrosis (CF), independent of documented infections or onset of pulmonary symptoms. New initiatives in clinical care are focusing on detection and characterization of preclinical disease. Thus, animal models are needed which recapitulate the pulmonary phenotype characteristic of early stage CF.
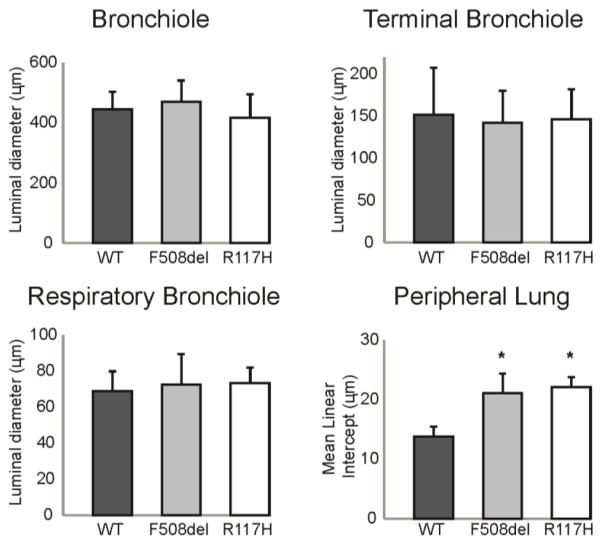
Methods: We investigated young CF mice to determine if they exhibit pulmonary pathophysiology consistent with the early CF lung phenotype. Lung histology and pulmonary mechanics were examined in 12- to 16-week-old congenic C57bl/6 F508del and R117H CF mice using a forced oscillation technique (flexiVent).
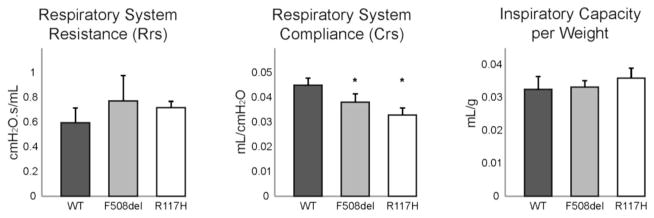
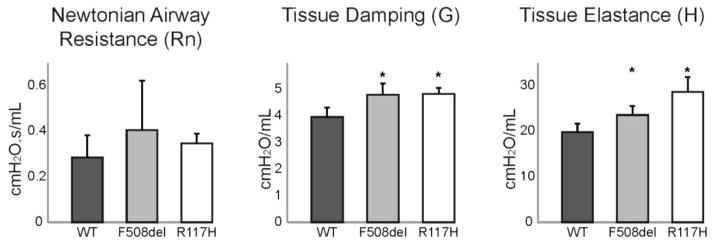
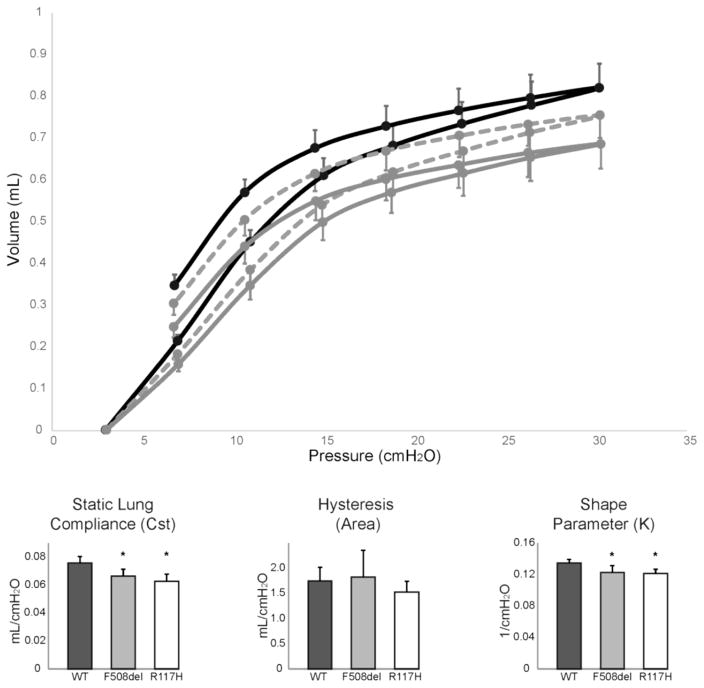
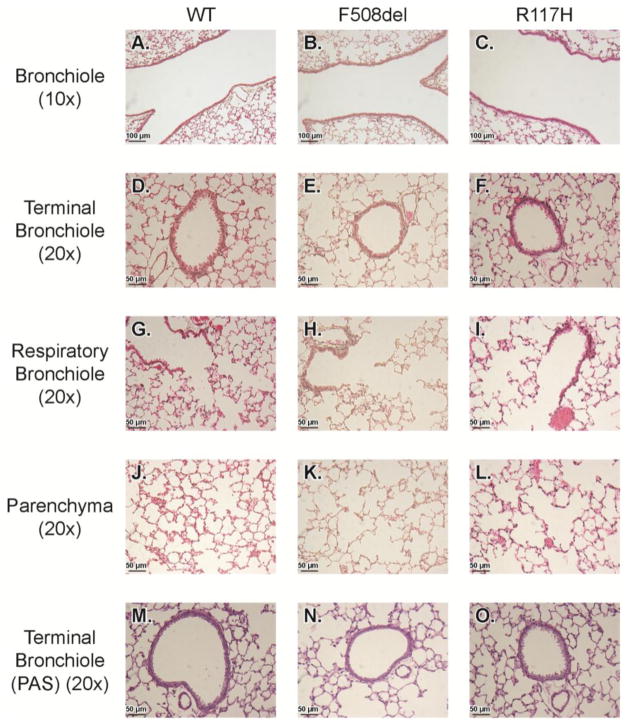
Results: There were no significant differences in the resistance of the large airways. However, in both CF mouse models, prominent differences in the mechanical properties of the peripheral lung compartment were identified including decreased static lung compliance, increased elastance and increased tissue damping. CF mice also had distal airspace enlargement with significantly increased mean linear intercept distances.
Conclusions: An impaired ability to stretch and expand the peripheral lung compartment, as well as increased distances between gas exchange surfaces, were present in young CF mice carrying two independent Cftr mutations. This altered pulmonary histopathophysiology in the peripheral lung compartment, which develops in the absence of infection, is similar to the early lung phenotype of CF patients.
Keywords: Cystic fibrosis; Lung mechanics; Mouse models.
Copyright © 2016 European Cystic Fibrosis Society. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Kozlowska WJ, Bush A, Wade A, Aurora P, Carr SB, Castle RA, Hoo AF, Lum S, Price J, Ranganathan S, Saunders C, Stanojevic S, Stroobant J, Wallis C, Stocks J London Cystic Fibrosis C. Lung function from infancy to the preschool years after clinical diagnosis of cystic fibrosis. American journal of respiratory and critical care medicine. 2008;178:42–49. - PubMed
-
- Linnane BM, Hall GL, Nolan G, Brennan S, Stick SM, Sly PD, Robertson CF, Robinson PJ, Franklin PJ, Turner SW, Ranganathan SC, Arest CF. Lung function in infants with cystic fibrosis diagnosed by newborn screening. American journal of respiratory and critical care medicine. 2008;178:1238–1244. - PubMed
-
- Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. American journal of respiratory and critical care medicine. 1995;151:1075–1082. - PubMed
-
- Hoo AF, Thia LP, Nguyen TT, Bush A, Chudleigh J, Lum S, Ahmed D, Balfour Lynn I, Carr SB, Chavasse RJ, Costeloe KL, Price J, Shankar A, Wallis C, Wyatt HA, Wade A, Stocks J London Cystic Fibrosis C. Lung function is abnormal in 3-month-old infants with cystic fibrosis diagnosed by newborn screening. Thorax. 2012;67:874–881. - PubMed
-
- Ramsey BW, Banks-Schlegel S, Accurso FJ, Boucher RC, Cutting GR, Engelhardt JF, Guggino WB, Karp CL, Knowles MR, Kolls JK, LiPuma JJ, Lynch S, McCray PB, Jr, Rubenstein RC, Singh PK, Sorscher E, Welsh M. Future directions in early cystic fibrosis lung disease research: An nhlbi workshop report. American journal of respiratory and critical care medicine. 2012;185:887–892. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
