

Insights into the Mechanisms of Basal Coordination of Transcription Using a Genome-Reduced Bacterium
- PMID: 27237741
- PMCID: PMC4920955
- DOI: 10.1016/j.cels.2016.04.015
Insights into the Mechanisms of Basal Coordination of Transcription Using a Genome-Reduced Bacterium
Erratum in
-
Insights into the Mechanisms of Basal Coordination of Transcription Using a Genome-Reduced Bacterium.Cell Syst. 2018 Aug 22;7(2):227-229. doi: 10.1016/j.cels.2018.08.006. Cell Syst. 2018. PMID: 30138582 Free PMC article. No abstract available.
Abstract
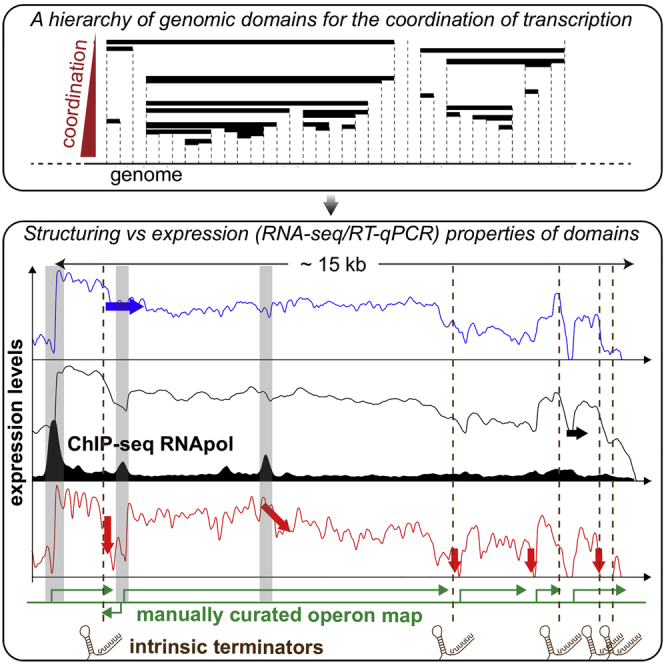
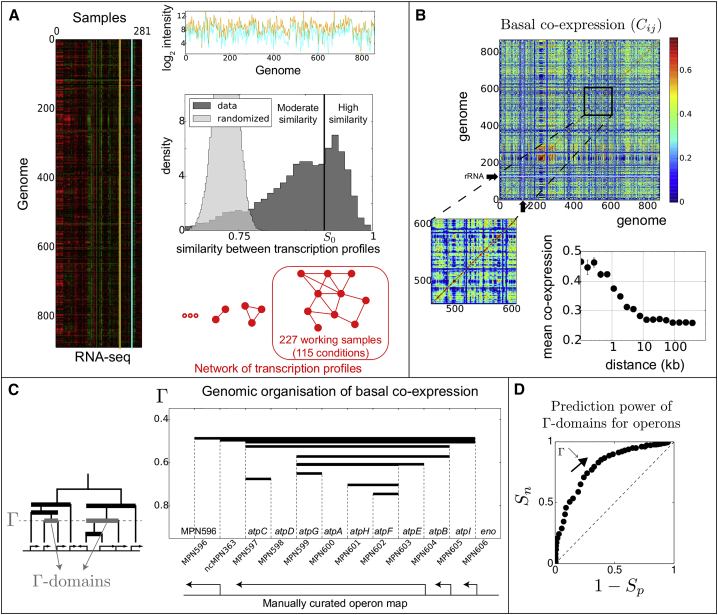
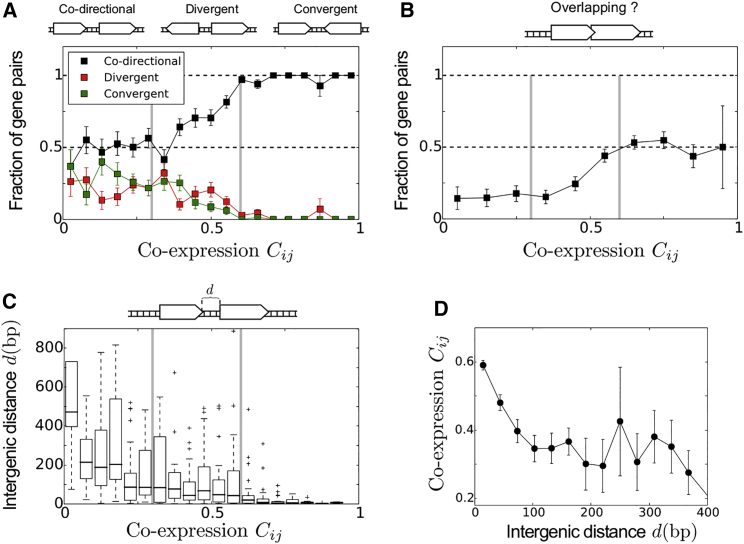
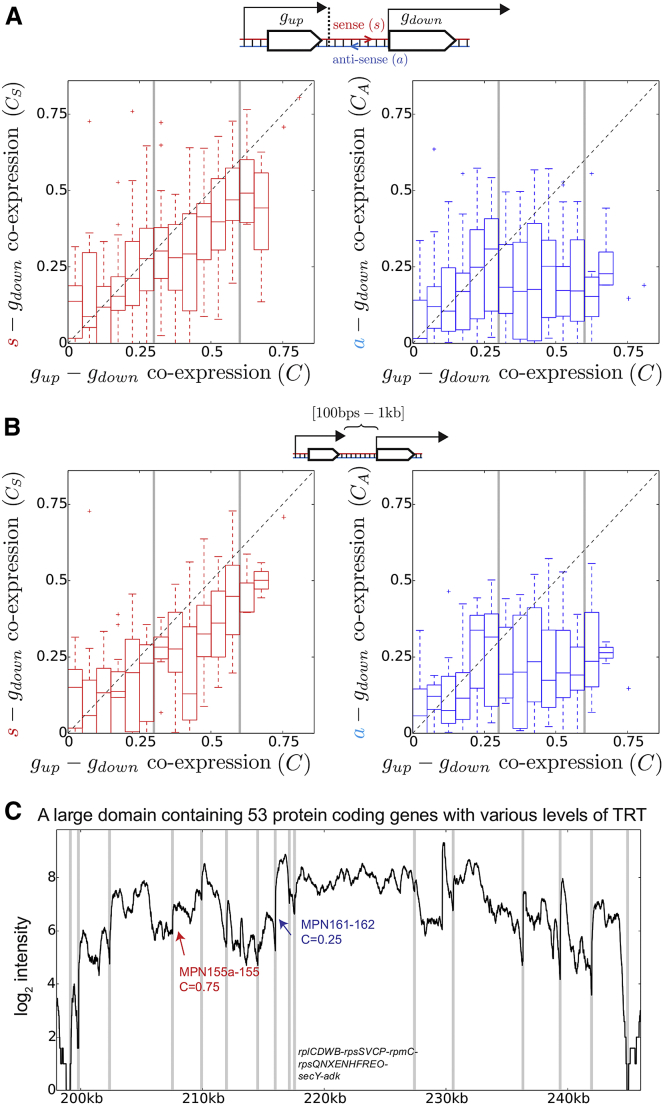
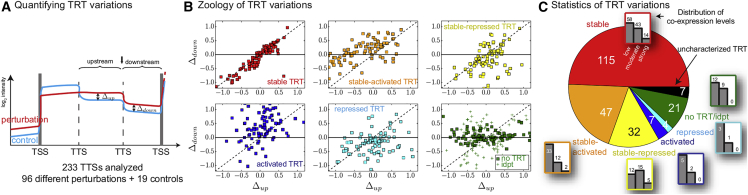
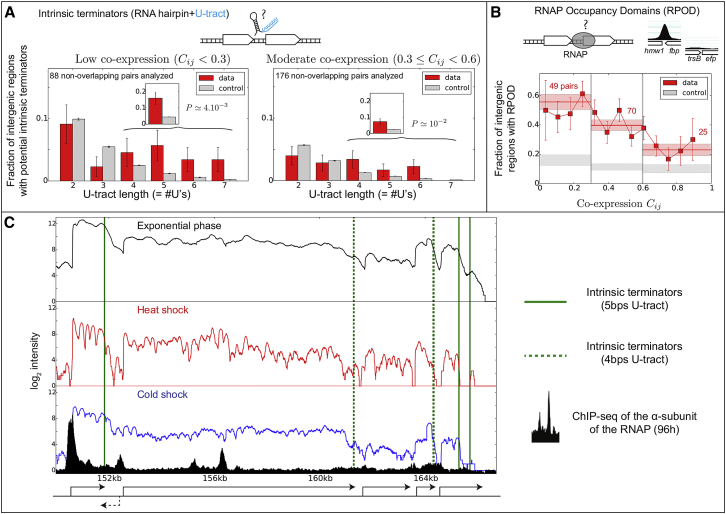
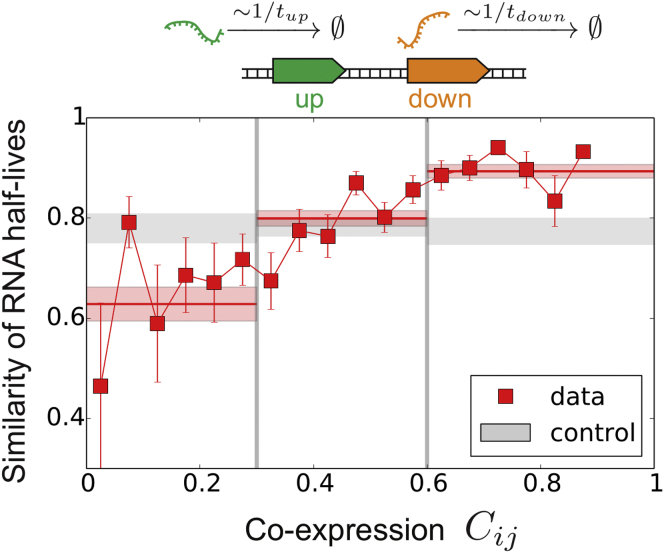
Coordination of transcription in bacteria occurs at supra-operonic scales, but the extent, specificity, and mechanisms of such regulation are poorly understood. Here, we tackle this problem by profiling the transcriptome of the model organism Mycoplasma pneumoniae across 115 growth conditions. We identify three qualitatively different levels of co-expression corresponding to distinct relative orientations and intergenic properties of adjacent genes. We reveal that the degree of co-expression between co-directional adjacent operons, and more generally between genes, is tightly related to their capacity to be transcribed en bloc into the same mRNA. We further show that this genome-wide pervasive transcription of adjacent genes and operons is specifically repressed by DNA regions preferentially bound by RNA polymerases, by intrinsic terminators, and by large intergenic distances. Taken together, our findings suggest that the basal coordination of transcription is mediated by the physical entities and mechanical properties of the transcription process itself, and that operon-like behaviors may strongly vary from condition to condition.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Chen Y.-J., Liu P., Nielsen A.A., Brophy J.A., Clancy K., Peterson T., Voigt C.A. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods. 2013;10:659–664. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
