

SIRT Is Required for EDP-Mediated Protective Responses toward Hypoxia-Reoxygenation Injury in Cardiac Cells
- PMID: 27242531
- PMCID: PMC4868841
- DOI: 10.3389/fphar.2016.00124
SIRT Is Required for EDP-Mediated Protective Responses toward Hypoxia-Reoxygenation Injury in Cardiac Cells
Abstract
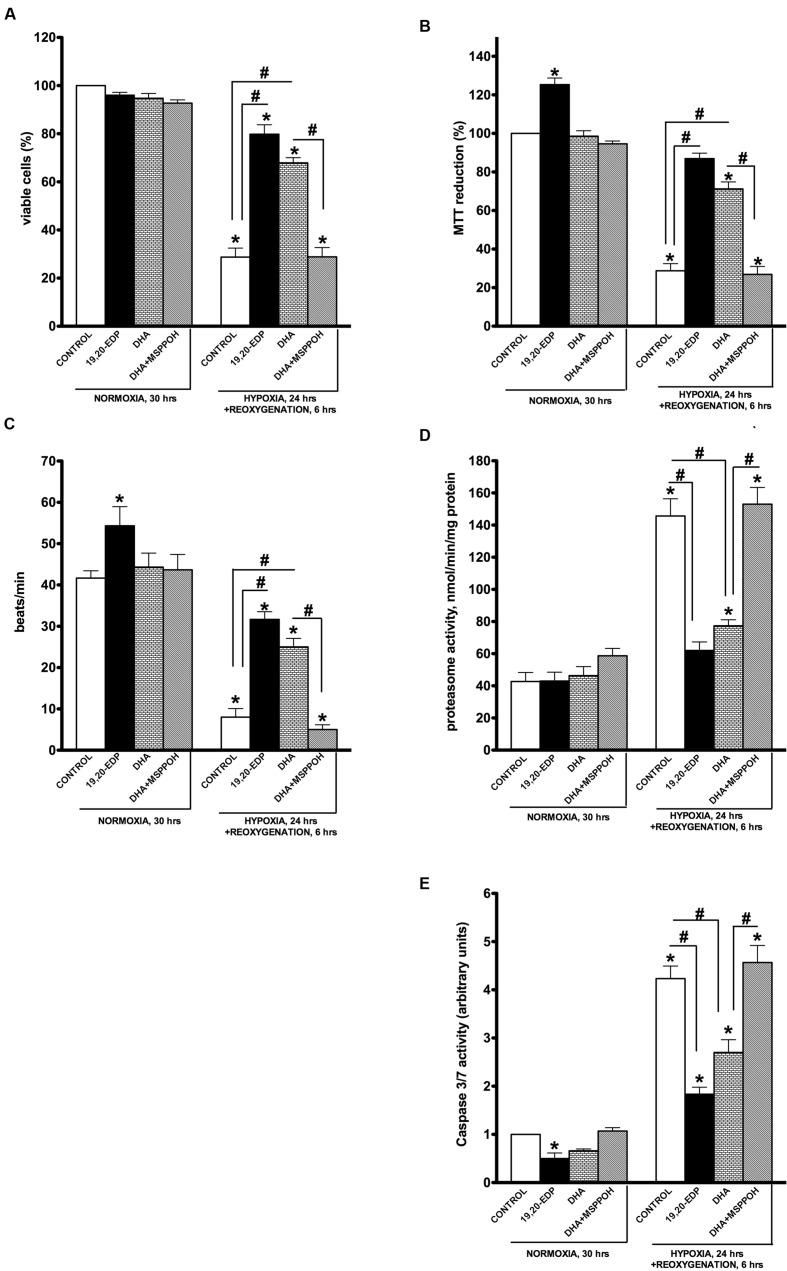
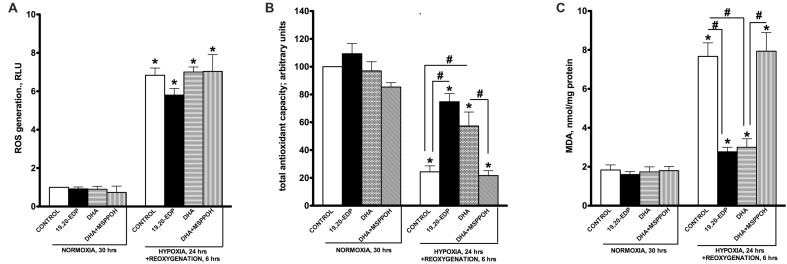
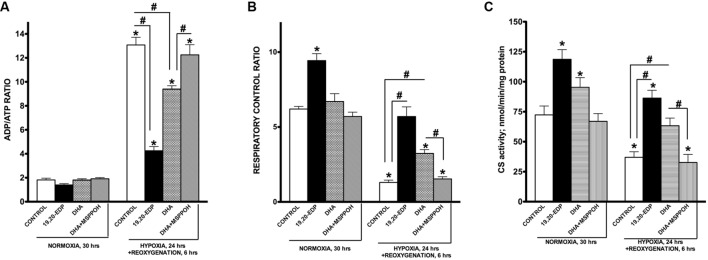
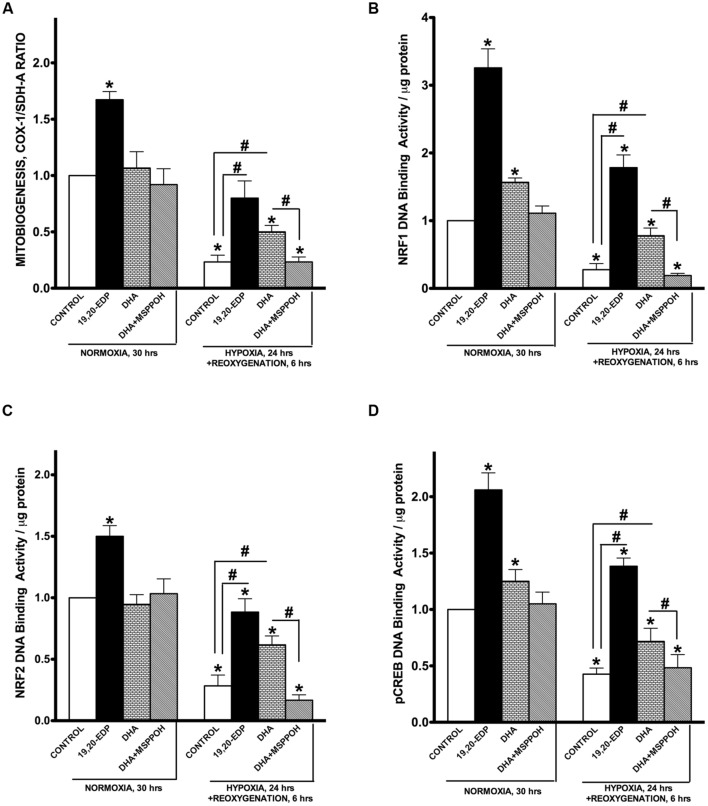
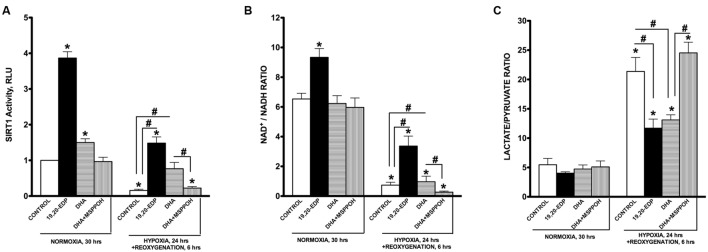
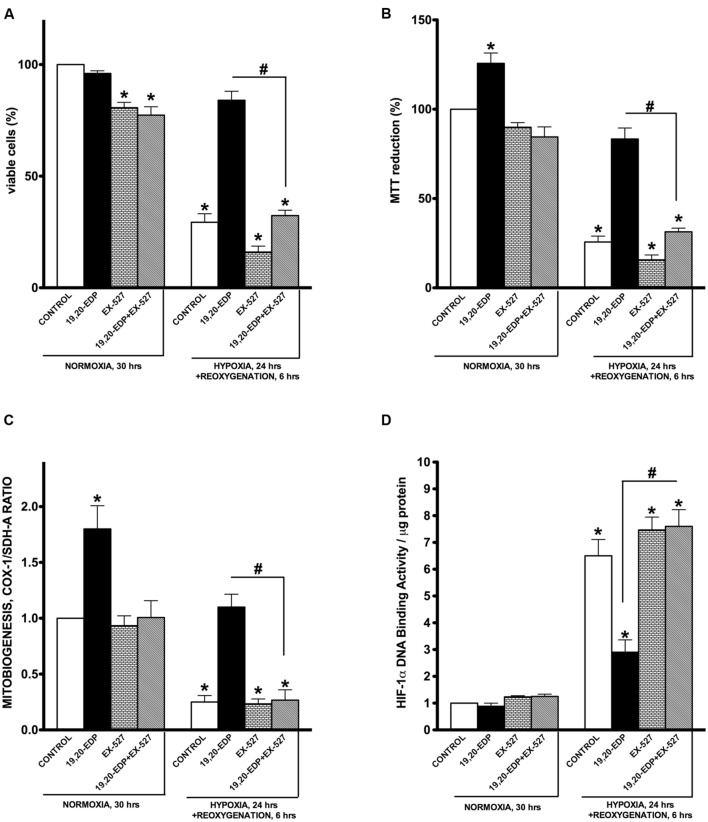
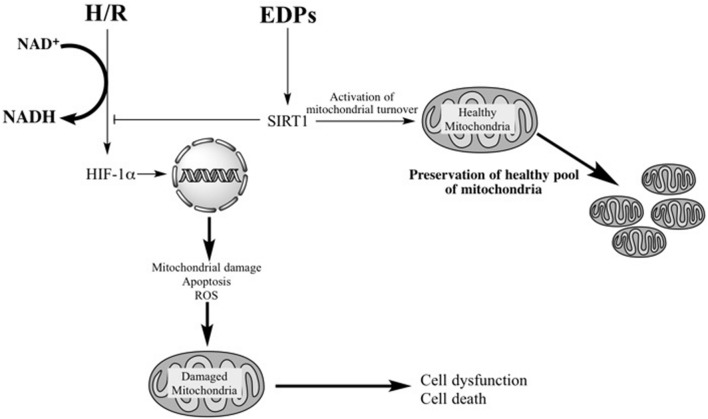
Hypoxia-reoxygenation (H/R) injury is known to cause extensive injury to cardiac myocardium promoting development of cardiac dysfunction. Despite the vast number of studies dedicated to studying H/R injury, the molecular mechanisms behind it are multiple, complex, and remain very poorly understood, which makes development of novel pharmacological agents challenging. Docosahexaenoic acid (DHA, 22:6n3) is an n - 3 polyunsaturated fatty acid obtained from dietary sources, which produces numerous effects including regulation of cell survival and death mechanisms. The beneficial effects of DHA toward the cardiovascular system are well documented but the relative role of DHA or one of its more potent metabolites is unresolved. Emerging evidence indicates that cytochrome P450 (CYP) epoxygenase metabolites of DHA, epoxydocosapentaenoic acids (EDPs), have more potent biological activity than DHA in cardiac cells. In this study we examined whether EDPs protect HL-1 cardiac cells from H/R injury. Our observations demonstrate that treatment with 19,20-EDP protected HL-1 cardiac cells from H/R damage through a mechanism(s) protecting and enhancing mitochondrial quality. EDP treatment increased the relative rates of mitobiogenesis and mitochondrial respiration in control and H/R exposed cardiac cells. The observed EDP protective response toward H/R injury involved SIRT1-dependent pathways.
Keywords: cardiac cells; docosahexaenoic acid; epoxydocosapentaenoic acids; hypoxia–reoxygenation; mitobiogenesis; mitochondrial function.
Figures
References
-
- Ayalew-Pervanchon A., Rousseau D., Moreau D., Assayag P., Weill P., Grynberg A. (2007). Long-term effect of dietary {alpha}-linolenic acid or decosahexaenoic acid on incorporation of decosahexaenoic acid in membranes and its influence on rat heart in vivo. Am. J. Physiol. Heart Circ. Physiol. 293 H2296–H2304. 10.1152/ajpheart.00194.2007 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources