

Tracking Strains in the Microbiome: Insights from Metagenomics and Models
- PMID: 27242733
- PMCID: PMC4871868
- DOI: 10.3389/fmicb.2016.00712
Tracking Strains in the Microbiome: Insights from Metagenomics and Models
Abstract
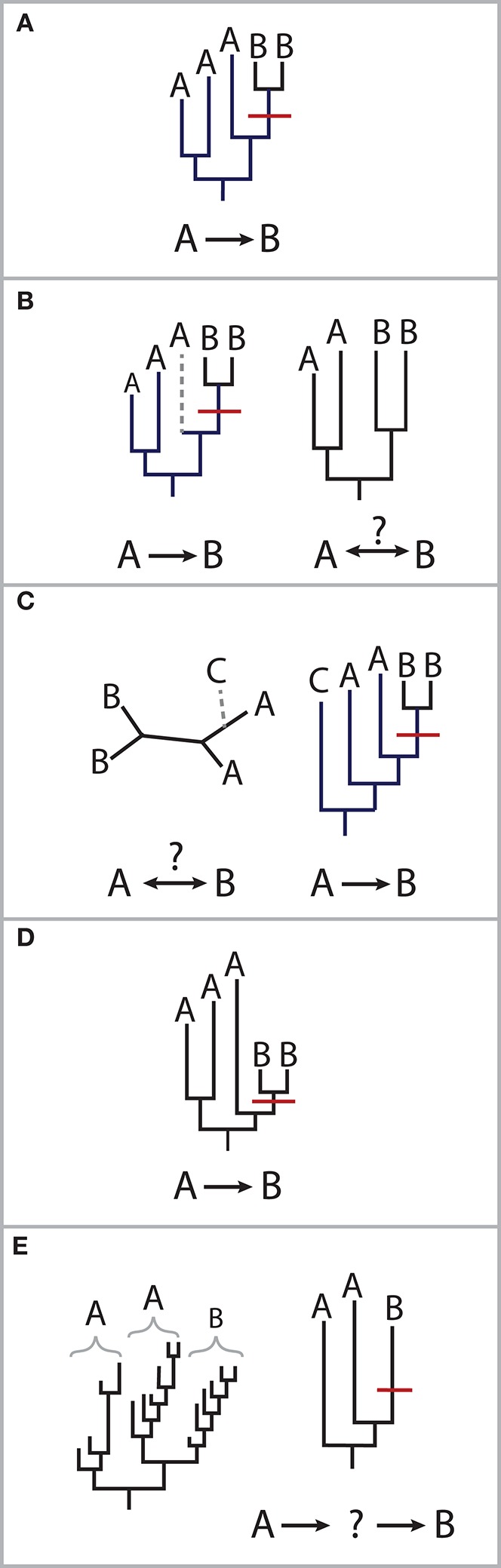
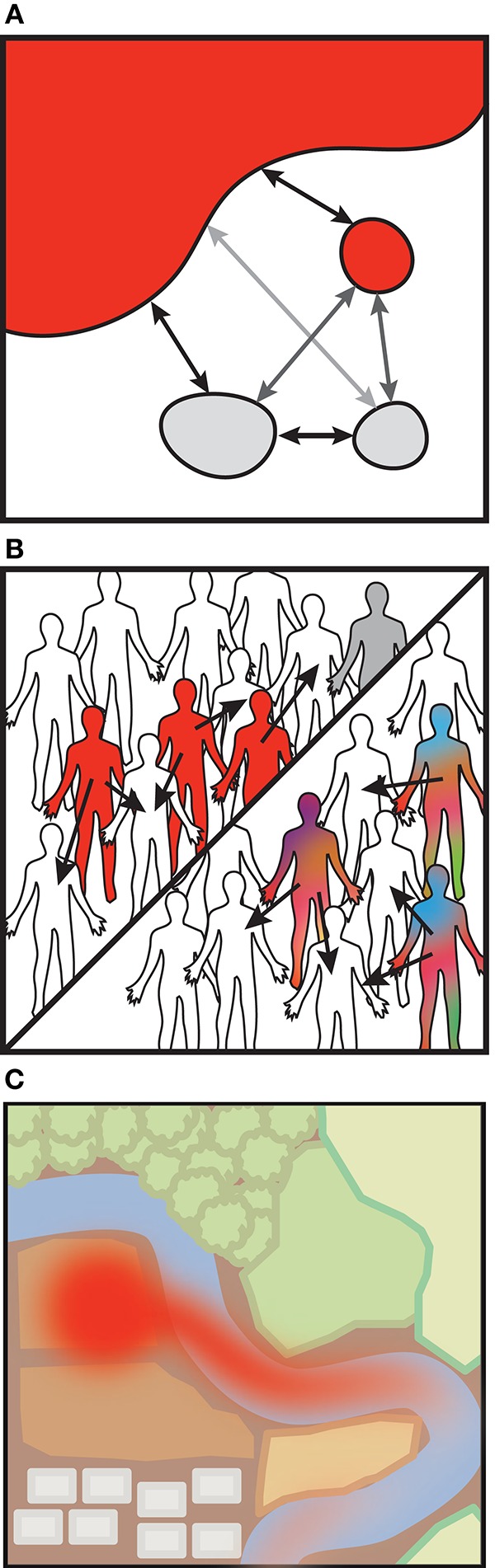
Transmission usually refers to the movement of pathogenic organisms. Yet, commensal microbes that inhabit the human body also move between individuals and environments. Surprisingly little is known about the transmission of these endogenous microbes, despite increasing realizations of their importance for human health. The health impacts arising from the transmission of commensal bacteria range widely, from the prevention of autoimmune disorders to the spread of antibiotic resistance genes. Despite this importance, there are outstanding basic questions: what is the fraction of the microbiome that is transmissible? What are the primary mechanisms of transmission? Which organisms are the most highly transmissible? Higher resolution genomic data is required to accurately link microbial sources (such as environmental reservoirs or other individuals) with sinks (such as a single person's microbiome). New computational advances enable strain-level resolution of organisms from shotgun metagenomic data, allowing the transmission of strains to be followed over time and after discrete exposure events. Here, we highlight the latest techniques that reveal strain-level resolution from raw metagenomic reads and new studies that are tracking strains across people and environments. We also propose how models of pathogenic transmission may be applied to study the movement of commensals between microbial communities.
Keywords: bacterial genomics; biological; genotyping techniques; metagenomics; microbiome; models; strain diversity.
Figures
References
-
- Anderson R. M., May R. M. (1979). Population biology of infectious diseases: part I. Nature 280, 361–367. - PubMed
-
- Benham B. L., Baffaut C., Zeckoski R. W., Mankin K. R., Pachepsky Y. A., Sadeghi A. M., et al. (2006). Modeling bacteria fate and transport in watersheds to support TMDLs. Trans. ASABE 49, 987–1002. 10.13031/2013.21739 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources