

Transcriptome Remodeling Contributes to Epidemic Disease Caused by the Human Pathogen Streptococcus pyogenes
- PMID: 27247229
- PMCID: PMC4895104
- DOI: 10.1128/mBio.00403-16
Transcriptome Remodeling Contributes to Epidemic Disease Caused by the Human Pathogen Streptococcus pyogenes
Abstract
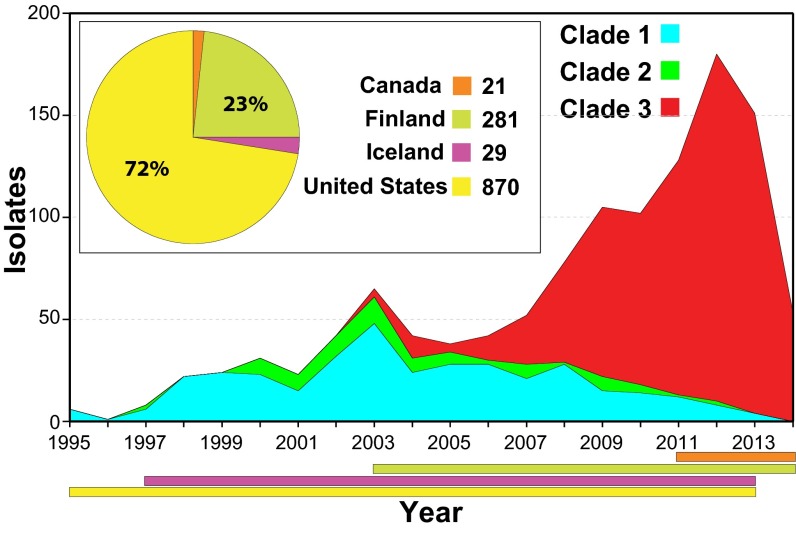
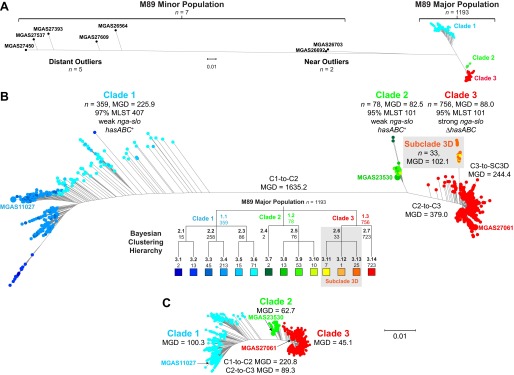
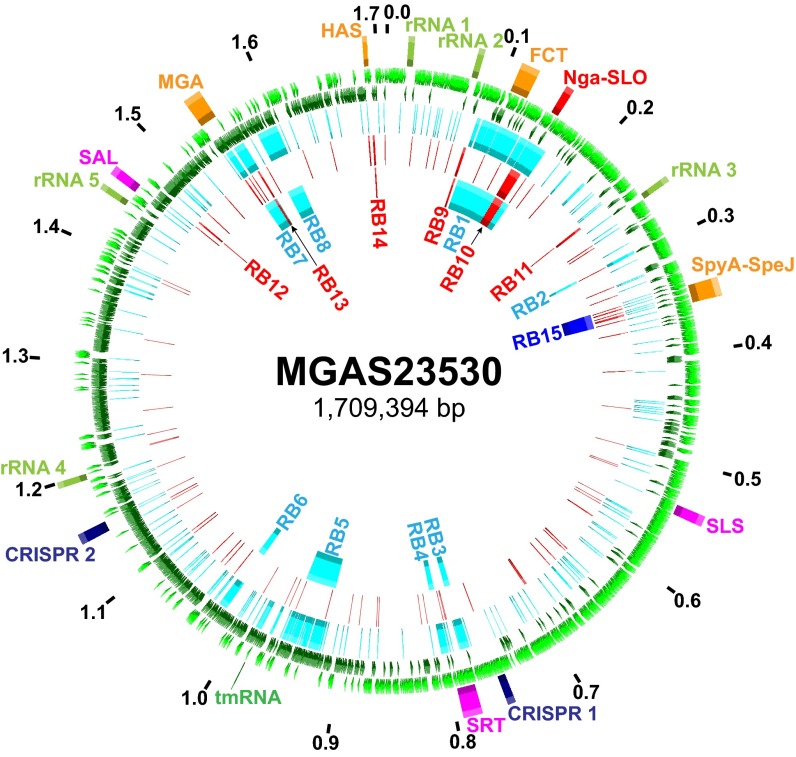
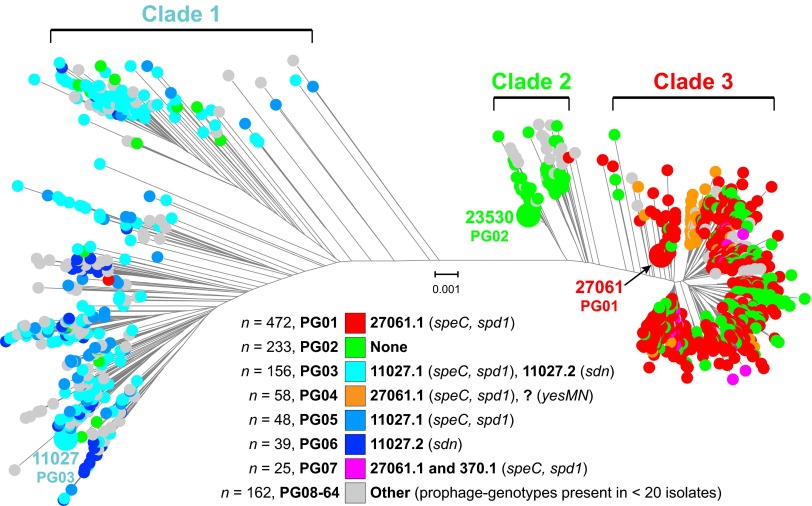
For over a century, a fundamental objective in infection biology research has been to understand the molecular processes contributing to the origin and perpetuation of epidemics. Divergent hypotheses have emerged concerning the extent to which environmental events or pathogen evolution dominates in these processes. Remarkably few studies bear on this important issue. Based on population pathogenomic analysis of 1,200 Streptococcus pyogenes type emm89 infection isolates, we report that a series of horizontal gene transfer events produced a new pathogenic genotype with increased ability to cause infection, leading to an epidemic wave of disease on at least two continents. In the aggregate, these and other genetic changes substantially remodeled the transcriptomes of the evolved progeny, causing extensive differential expression of virulence genes and altered pathogen-host interaction, including enhanced immune evasion. Our findings delineate the precise molecular genetic changes that occurred and enhance our understanding of the evolutionary processes that contribute to the emergence and persistence of epidemically successful pathogen clones. The data have significant implications for understanding bacterial epidemics and for translational research efforts to blunt their detrimental effects.
Importance: The confluence of studies of molecular events underlying pathogen strain emergence, evolutionary genetic processes mediating altered virulence, and epidemics is in its infancy. Although understanding these events is necessary to develop new or improved strategies to protect health, surprisingly few studies have addressed this issue, in particular, at the comprehensive population genomic level. Herein we establish that substantial remodeling of the transcriptome of the human-specific pathogen Streptococcus pyogenes by horizontal gene flow and other evolutionary genetic changes is a central factor in precipitating and perpetuating epidemic disease. The data unambiguously show that the key outcome of these molecular events is evolution of a new, more virulent pathogenic genotype. Our findings provide new understanding of epidemic disease.
Copyright © 2016 Beres et al.
Figures

References
-
- Caugant DA, Frøholm LO, Bøvre K, Holten E, Frasch CE, Mocca LF, Zollinger WD, Selander RK. 1986. Intercontinental spread of a genetically distinctive complex of clones of Neisseria meningitidis causing epidemic disease. Proc Natl Acad Sci U S A 83:4927–4931. doi:10.1073/pnas.83.13.4927. - DOI - PMC - PubMed
-
- Chewapreecha C, Harris SR, Croucher NJ, Turner C, Marttinen P, Cheng L, Pessia A, Aanensen DM, Mather AE, Page AJ, Salter SJ, Harris D, Nosten F, Goldblatt D, Corander J, Parkhill J, Turner P, Bentley SD. 2014. Dense genomic sampling identifies highways of pneumococcal recombination. Nat Genet 46:305–309. doi:10.1038/ng.2895. - DOI - PMC - PubMed
-
- Cui Y, Yu C, Yan Y, Li D, Li Y, Jombart T, Weinert LA, Wang Z, Guo Z, Xu L, Zhang Y, Zheng H, Qin N, Xiao X, Wu M, Wang X, Zhou D, Qi Z, Du Z, Wu H, Yang X, Cao H, Wang H, Wang J, Yao S, Rakin A, Li Y, Falush D, Balloux F, Achtman M, Song Y, Wang J, Yang R. 2013. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci U S A 110:577–582. doi:10.1073/pnas.1205750110. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
