

Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage
- PMID: 27247391
- PMCID: PMC4914194
- DOI: 10.1073/pnas.1606460113
Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage
Abstract
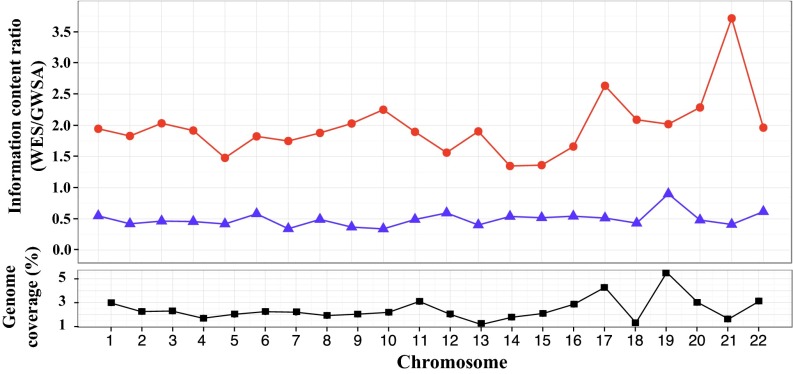
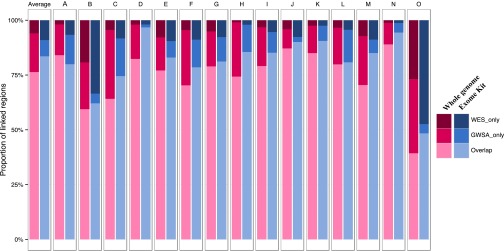
Principal component analysis (PCA), homozygosity rate estimations, and linkage studies in humans are classically conducted through genome-wide single-nucleotide variant arrays (GWSA). We compared whole-exome sequencing (WES) and GWSA for this purpose. We analyzed 110 subjects originating from different regions of the world, including North Africa and the Middle East, which are poorly covered by public databases and have high consanguinity rates. We tested and applied a number of quality control (QC) filters. Compared with GWSA, we found that WES provided an accurate prediction of population substructure using variants with a minor allele frequency > 2% (correlation = 0.89 with the PCA coordinates obtained by GWSA). WES also yielded highly reliable estimates of homozygosity rates using runs of homozygosity with a 1,000-kb window (correlation = 0.94 with the estimates provided by GWSA). Finally, homozygosity mapping analyses in 15 families including a single offspring with high homozygosity rates showed that WES provided 51% less genome-wide linkage information than GWSA overall but 97% more information for the coding regions. At the genome-wide scale, 76.3% of linked regions were found by both GWSA and WES, 17.7% were found by GWSA only, and 6.0% were found by WES only. For coding regions, the corresponding percentages were 83.5%, 7.4%, and 9.1%, respectively. With appropriate QC filters, WES can be used for PCA and adjustment for population substructure, estimating homozygosity rates in individuals, and powerful linkage analyses, particularly in coding regions.
Keywords: exome sequencing; genotyping array; homozygosity mapping; linkage analysis; population structure.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
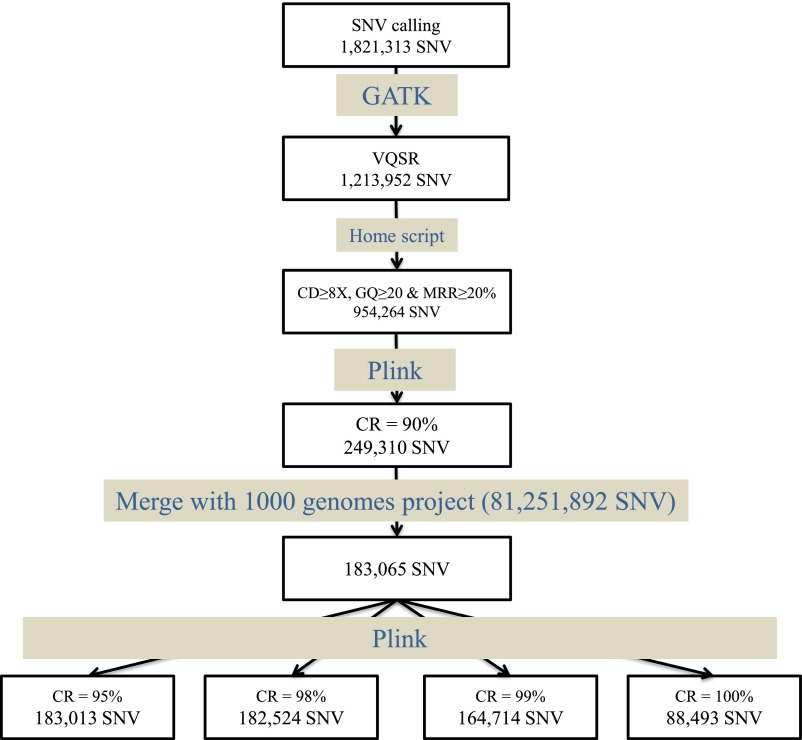
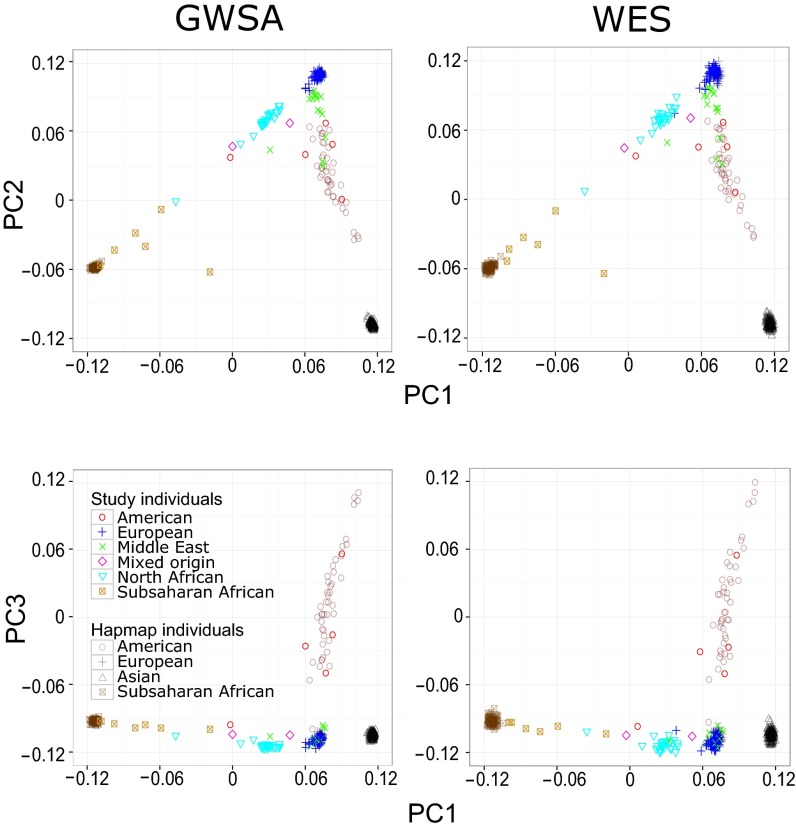
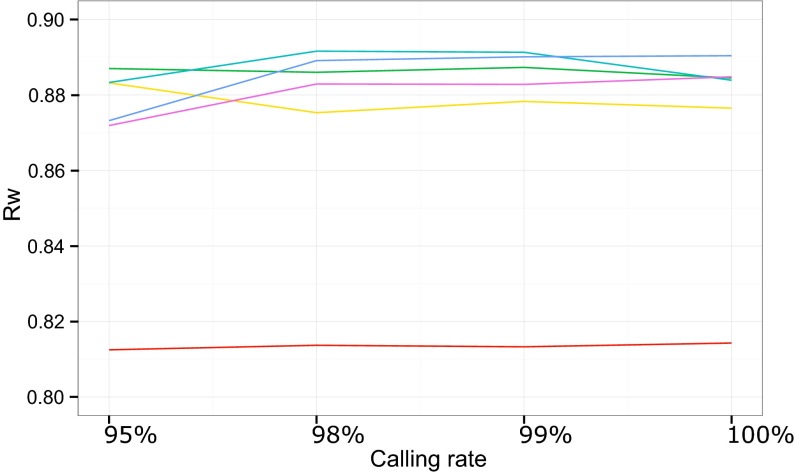
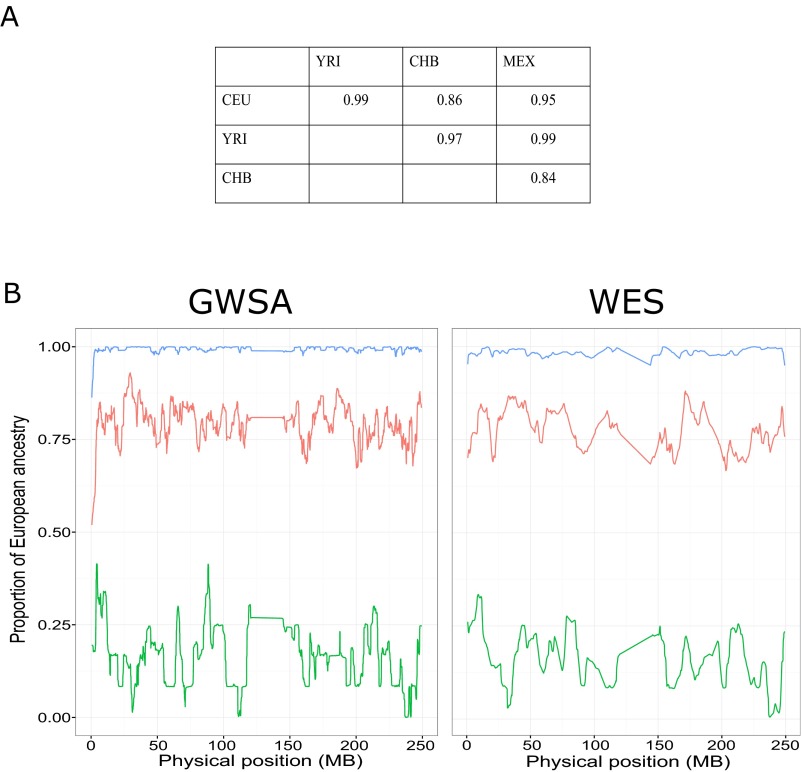
References
-
- Bamshad MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–755. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
