

Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism
- PMID: 27248322
- PMCID: PMC5190055
- DOI: 10.18632/oncotarget.9666
Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism
Abstract
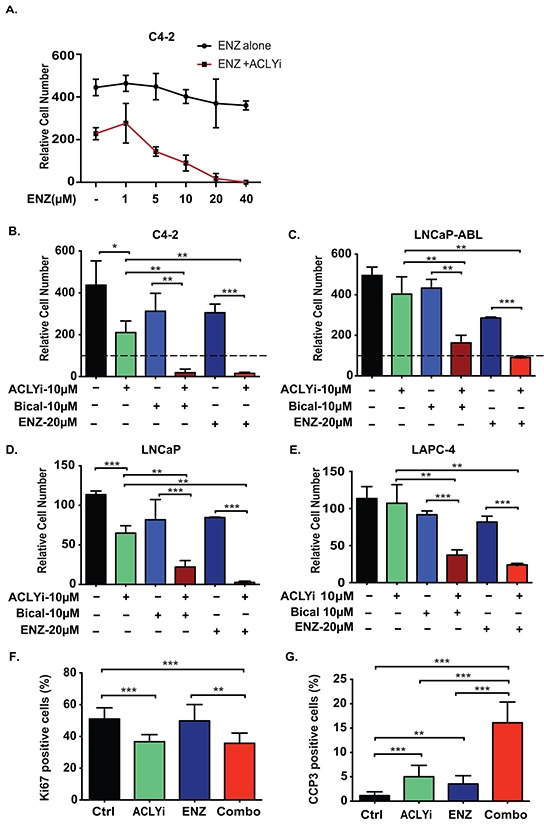
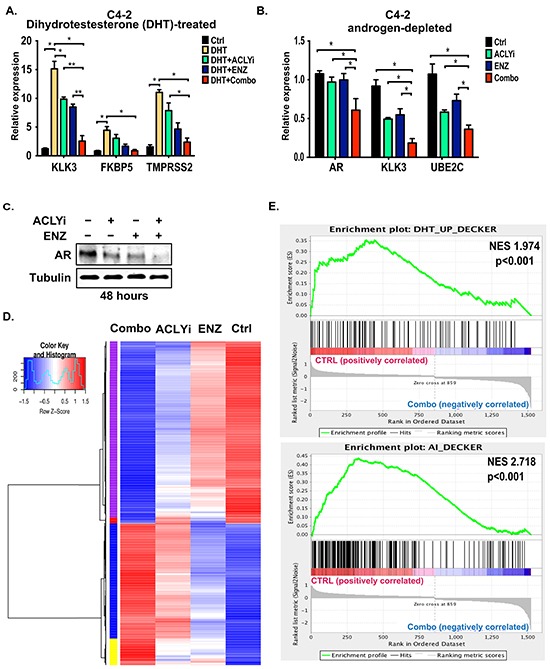
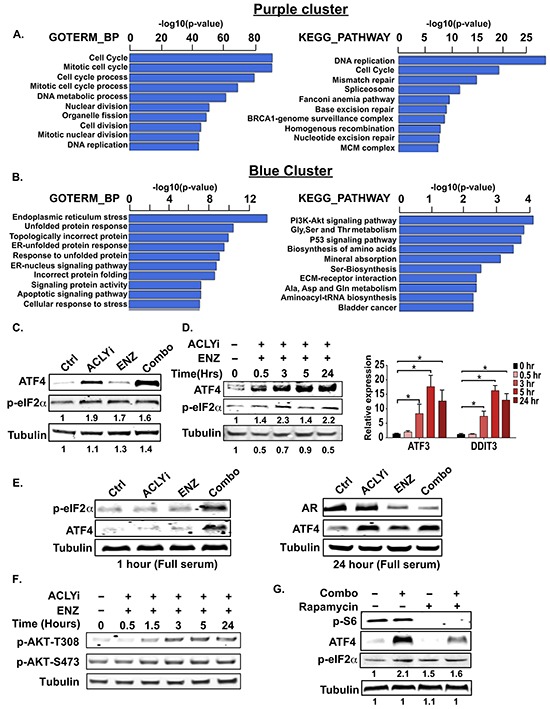
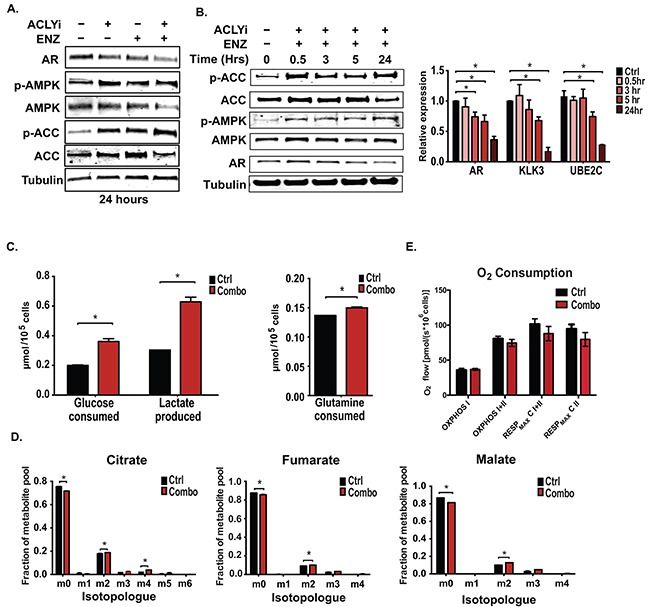
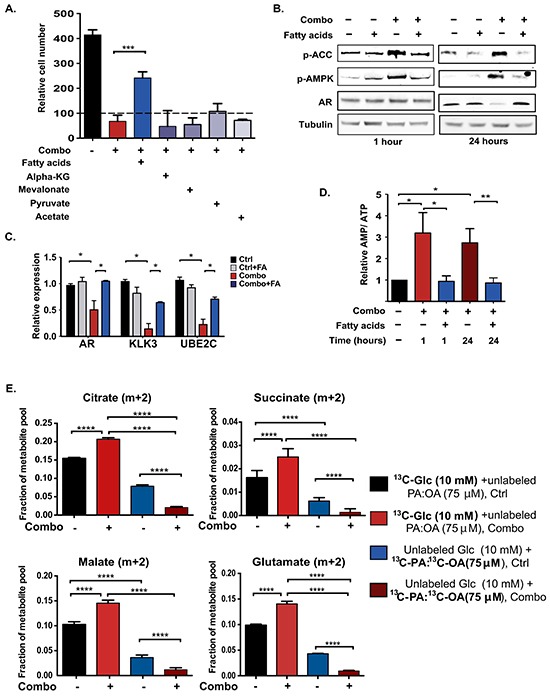
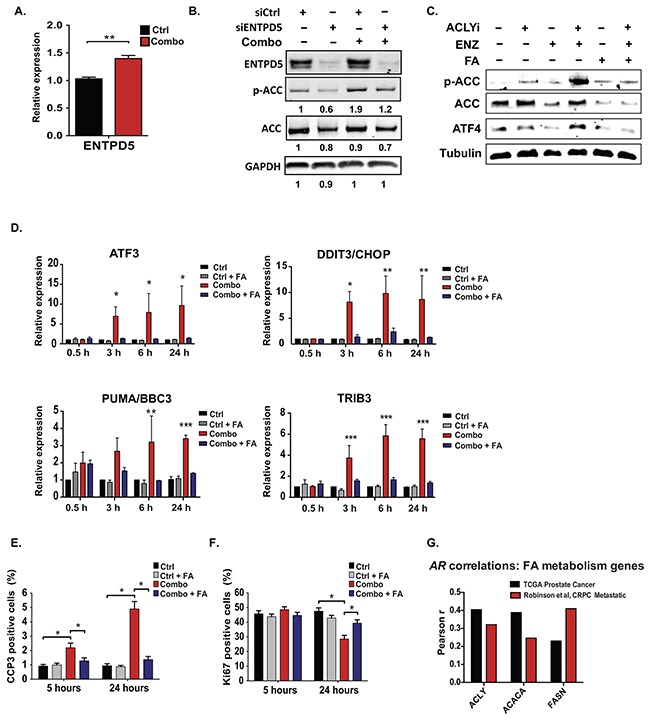
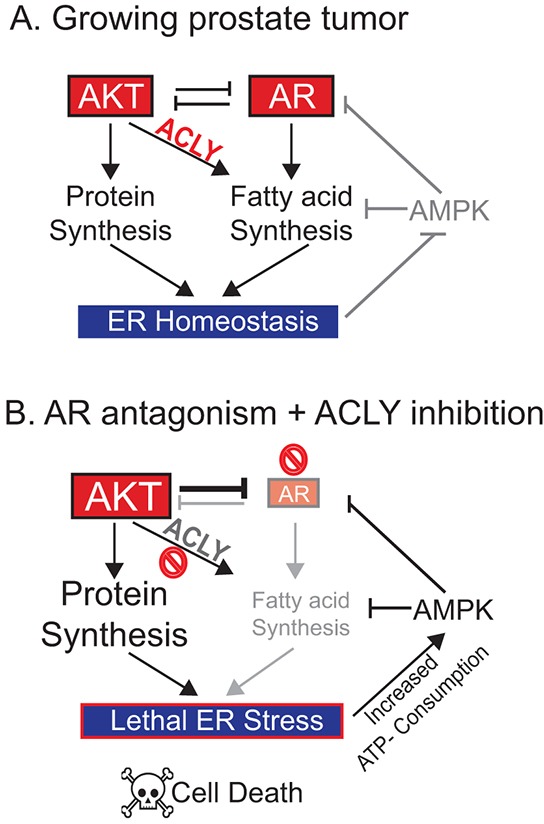
The androgen receptor (AR) plays a central role in prostate tumor growth. Inappropriate reactivation of the AR after androgen deprivation therapy promotes development of incurable castration-resistant prostate cancer (CRPC). In this study, we provide evidence that metabolic features of prostate cancer cells can be exploited to sensitize CRPC cells to AR antagonism. We identify a feedback loop between ATP-citrate lyase (ACLY)-dependent fatty acid synthesis, AMPK, and the AR in prostate cancer cells that could contribute to therapeutic resistance by maintaining AR levels. When combined with an AR antagonist, ACLY inhibition in CRPC cells promotes energetic stress and AMPK activation, resulting in further suppression of AR levels and target gene expression, inhibition of proliferation, and apoptosis. Supplying exogenous fatty acids can restore energetic homeostasis; however, this rescue does not occur through increased β-oxidation to support mitochondrial ATP production. Instead, concurrent inhibition of ACLY and AR may drive excess ATP consumption as cells attempt to cope with endoplasmic reticulum (ER) stress, which is prevented by fatty acid supplementation. Thus, fatty acid metabolism plays a key role in coordinating ER and energetic homeostasis in CRPC cells, thereby sustaining AR action and promoting proliferation. Consistent with a role for fatty acid metabolism in sustaining AR levels in prostate cancer in vivo, AR mRNA levels in human prostate tumors correlate positively with expression of ACLY and other fatty acid synthesis genes. The ACLY-AMPK-AR network can be exploited to sensitize CRPC cells to AR antagonism, suggesting novel therapeutic opportunities for prostate cancer.
Keywords: AMPK; ER stress; acetyl-CoA; fatty acid metabolism; prostate cancer.
Conflict of interest statement
Authors have no conflicts of interest to declare.
Figures
References
-
- Cancer Facts and Figures 2015. American Cancer Society 2015
-
- Mills IG. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat Rev Cancer. 2014;14:187–198. - PubMed
-
- Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG, Neal DE. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35–47. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
