

Protein Regulation in Signal Transduction
- PMID: 27252361
- PMCID: PMC4888820
- DOI: 10.1101/cshperspect.a005918
Protein Regulation in Signal Transduction
Abstract
SUMMARYCells must respond to a diverse, complex, and ever-changing mix of signals, using a fairly limited set of parts. Changes in protein level, protein localization, protein activity, and protein-protein interactions are critical aspects of signal transduction, allowing cells to respond highly specifically to a nearly limitless set of cues and also to vary the sensitivity, duration, and dynamics of the response. Signal-dependent changes in levels of gene expression and protein synthesis play an important role in regulation of protein levels, whereas posttranslational modifications of proteins regulate their degradation, localization, and functional interactions. Protein ubiquitylation, for example, can direct proteins to the proteasome for degradation or provide a signal that regulates their interactions and/or location within the cell. Similarly, protein phosphorylation by specific kinases is a key mechanism for augmenting protein activity and relaying signals to other proteins that possess domains that recognize the phosphorylated residues.
Copyright © 2016 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures





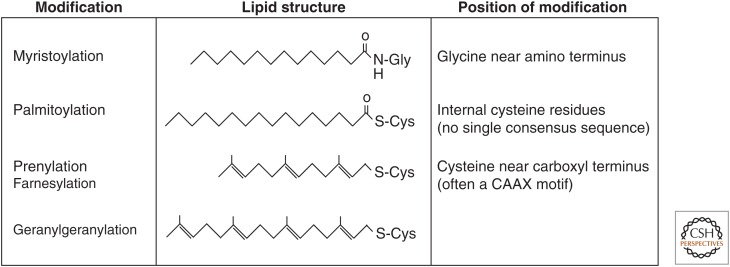

References
-
- Alexander J, Lim D, Joughin BA, Hegemann B, Hutchins JRA, Ehrenberger T, Ivins F, Sessa F, Hudecz O, Nigg EA, et al. 2011. Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci Signal 4: ra42. - PMC - PubMed
-
- Ambros V. 2001. microRNAs: Tiny regulators with great potential. Cell 107: 823–826. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources