

Rapid Proteasomal Degradation of Mutant Proteins Is the Primary Mechanism Leading to Tumorigenesis in Patients With Missense AIP Mutations
- PMID: 27253664
- PMCID: PMC4971335
- DOI: 10.1210/jc.2016-1307
Rapid Proteasomal Degradation of Mutant Proteins Is the Primary Mechanism Leading to Tumorigenesis in Patients With Missense AIP Mutations
Abstract
Context: The pathogenic effect of mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene (AIPmuts) in pituitary adenomas is incompletely understood. We have identified the primary mechanism of loss of function for missense AIPmuts.
Objective: This study sought to analyze the mechanism/speed of protein turnover of wild-type and missense AIP variants, correlating protein half-life with clinical parameters.
Design and setting: Half-life and protein-protein interaction experiments and cross-sectional analysis of AIPmut positive patients' data were performed in a clinical academic research institution.
Patients: Data were obtained from our cohort of pituitary adenoma patients and literature-reported cases.
Interventions: Protein turnover of endogenous AIP in two cell lines and fifteen AIP variants overexpressed in HEK293 cells was analyzed via cycloheximide chase and proteasome inhibition. Glutathione-S-transferase pull-down and quantitative mass spectrometry identified proteins involved in AIP degradation; results were confirmed by coimmunoprecipitation and gene knockdown. Relevant clinical data was collected.
Main outcome measures: Half-life of wild-type and mutant AIP proteins and its correlation with clinical parameters.
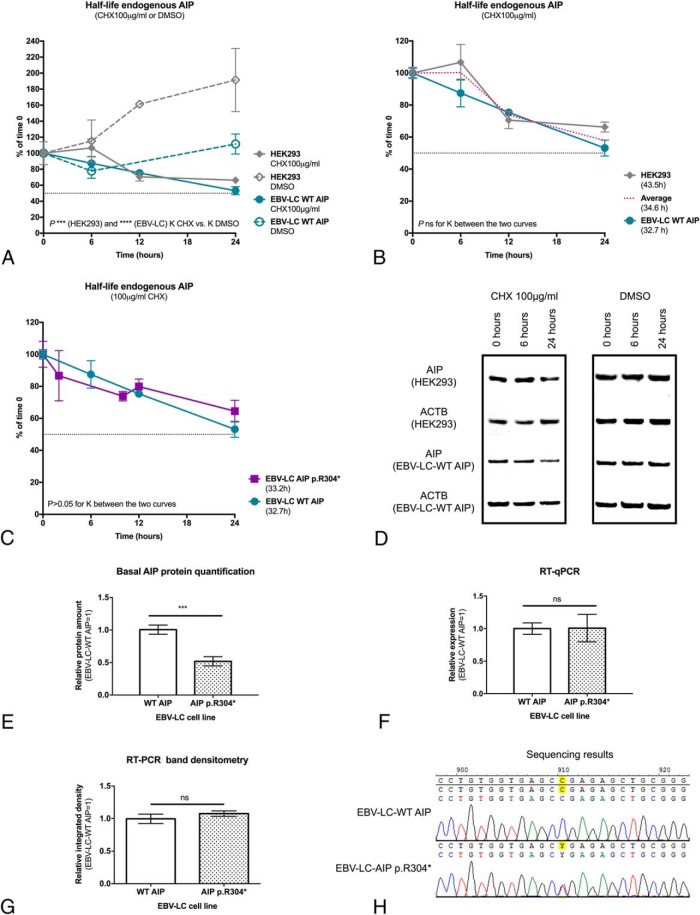
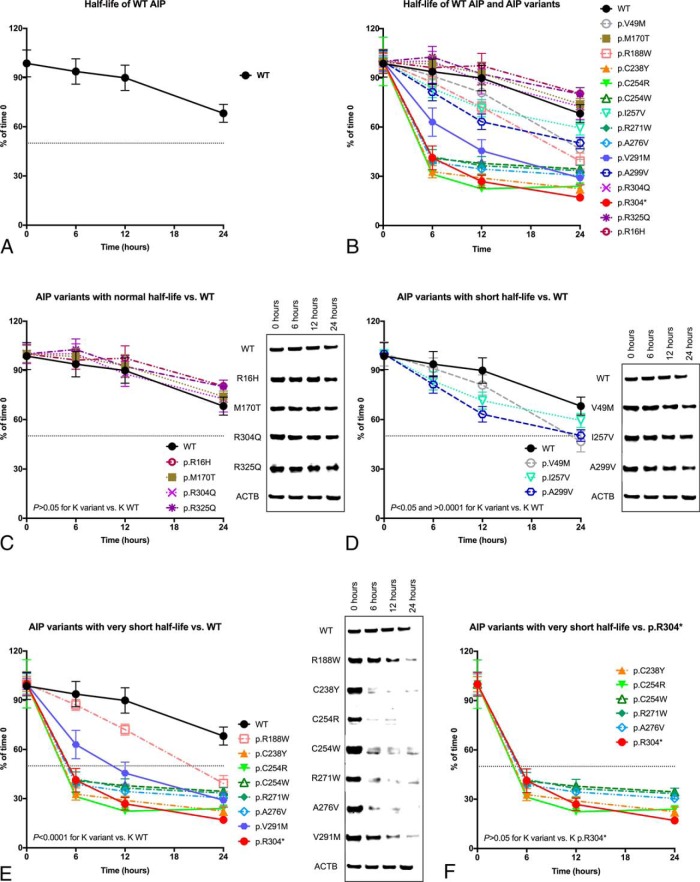
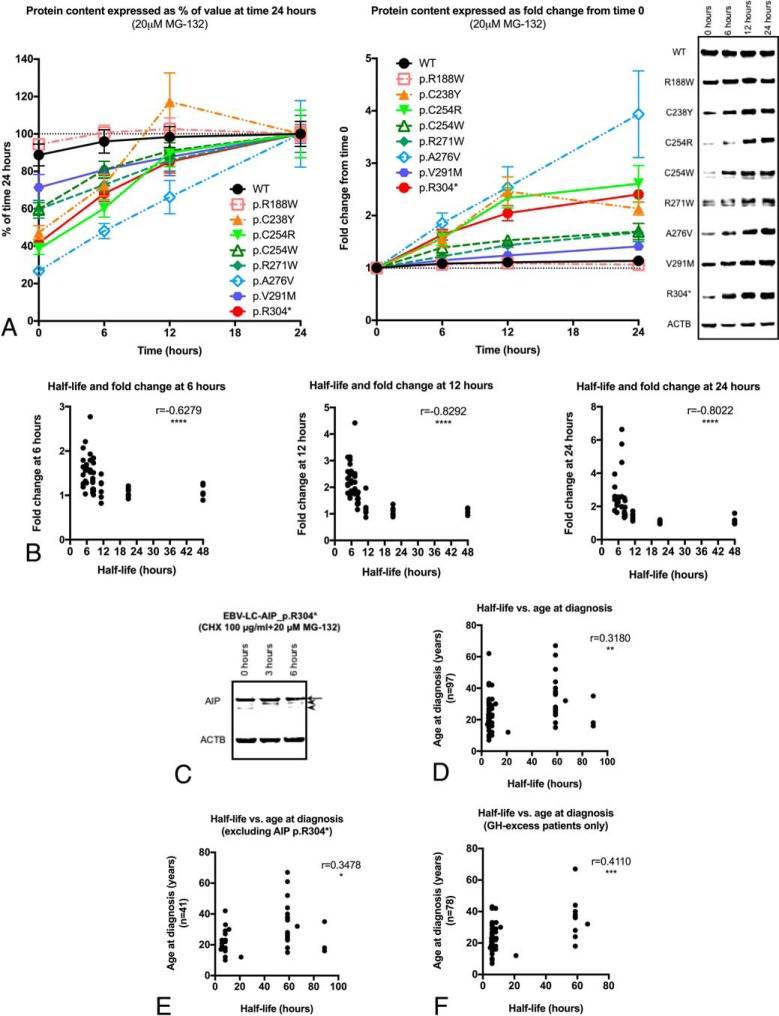
Results: Endogenous AIP half-life was similar in HEK293 and lymphoblastoid cells (43.5 and 32.7 h). AIP variants were divided into stable proteins (median, 77.7 h; interquartile range [IQR], 60.7-92.9 h), and those with short (median, 27 h; IQR, 21.6-28.7 h) or very short (median, 7.7 h; IQR, 5.6-10.5 h) half-life; proteasomal inhibition rescued the rapid degradation of mutant proteins. The experimental half-life significantly correlated with age at diagnosis of acromegaly/gigantism (r = 0.411; P = .002). The FBXO3-containing SKP1-CUL1-F-box protein complex was identified as the E3 ubiquitin-ligase recognizing AIP.
Conclusions: AIP is a stable protein, driven to ubiquitination by the SKP1-CUL1-F-box protein complex. Enhanced proteasomal degradation is a novel pathogenic mechanism for AIPmuts, with direct implications for the phenotype.
Trial registration: ClinicalTrials.gov NCT00461188.
Figures
References
-
- Vierimaa O, Georgitsi M, Lehtonen R, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312:1228–1230. - PubMed
-
- Daly AF, Vanbellinghen JF, Khoo SK, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: Analysis in 73 families. J Clin Endocrinol Metab. 2007;92:1891–1896. - PubMed
-
- Leontiou CA, Gueorguiev M, van der Spuy J, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab. 2008;93:2390–2401. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
