

Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease
- PMID: 27259053
- PMCID: PMC4908191
- DOI: 10.1016/j.ajhg.2016.05.004
Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease
Abstract
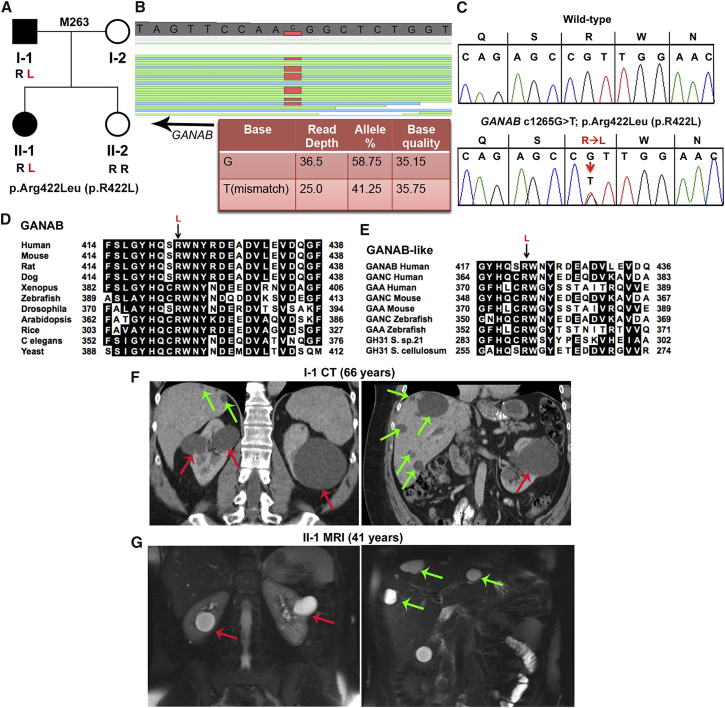
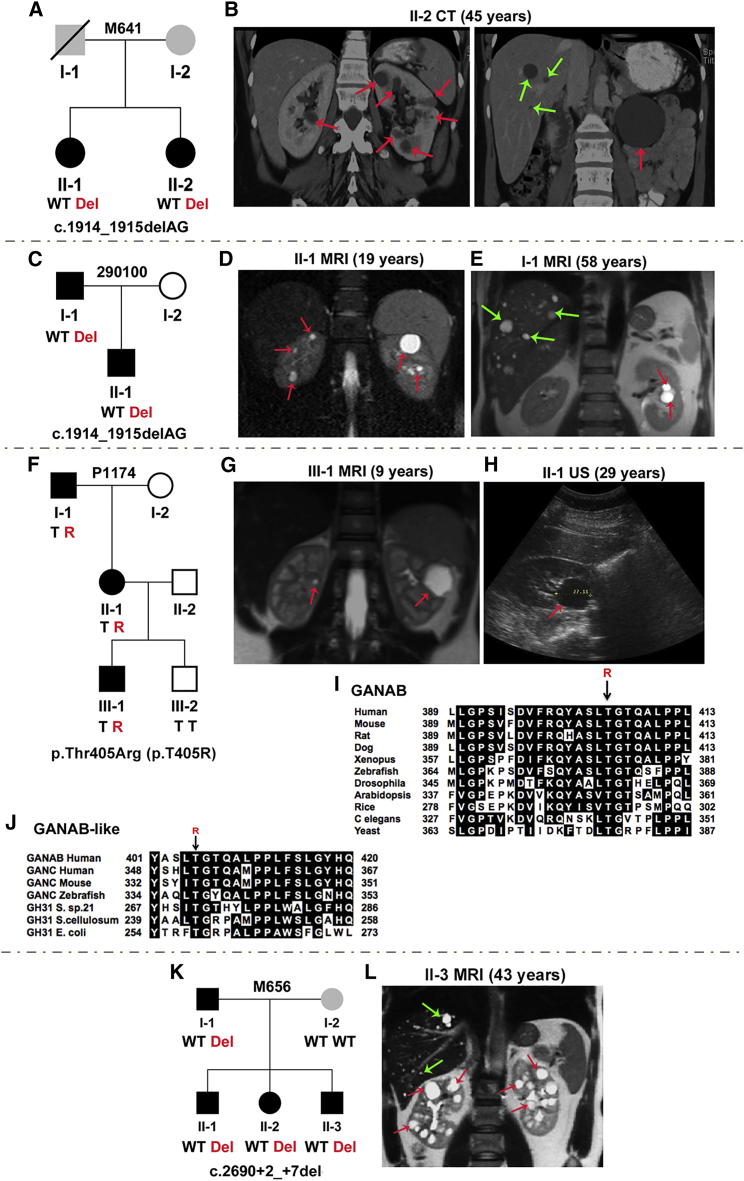
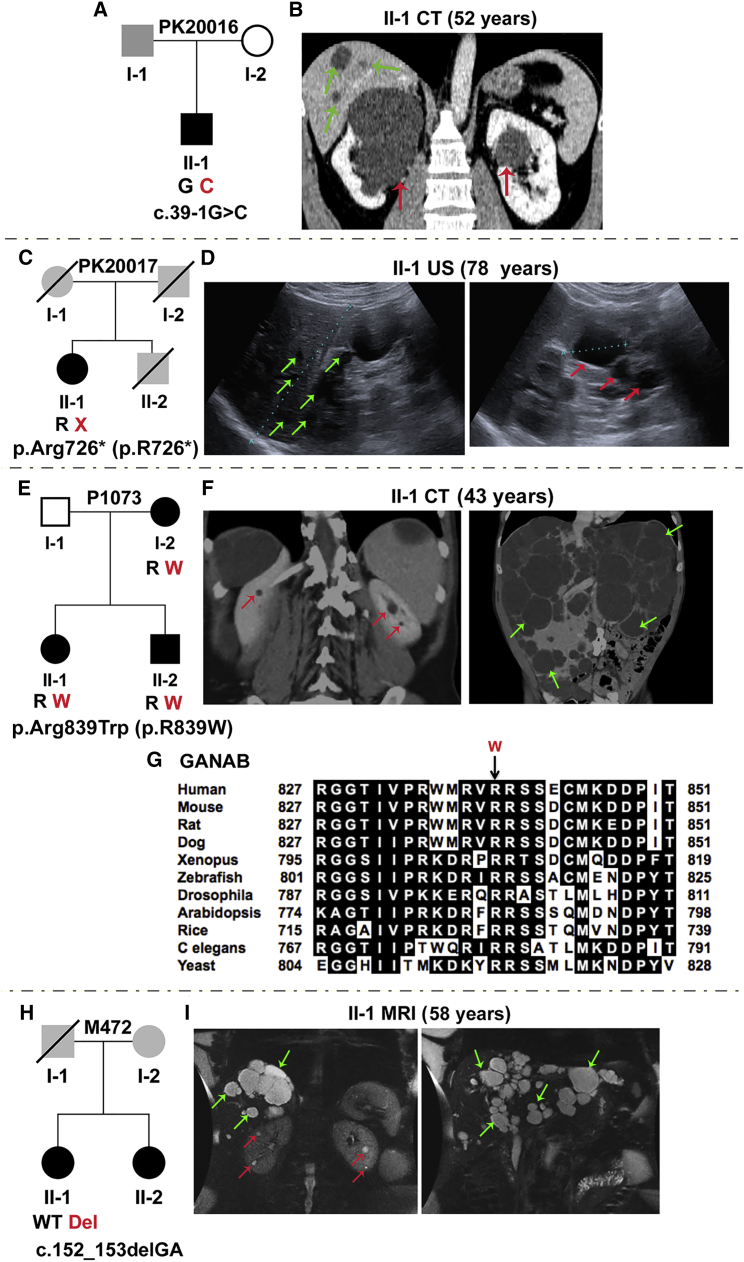
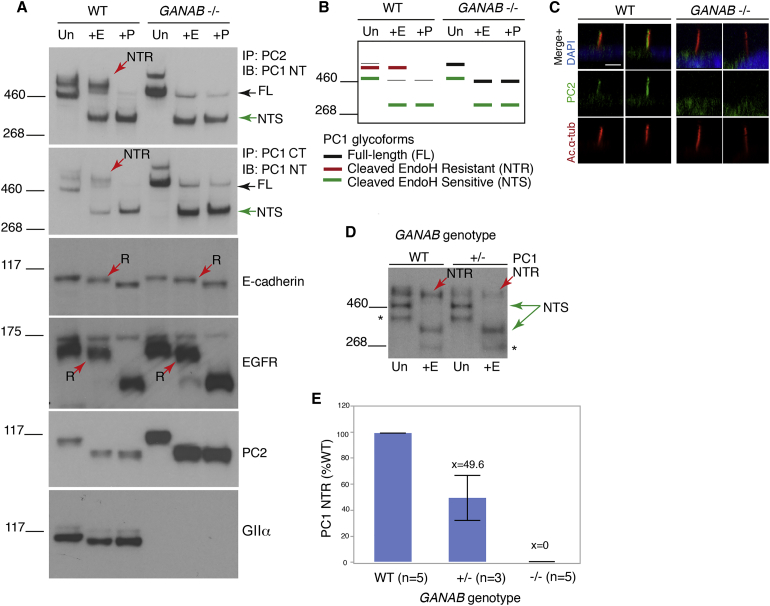
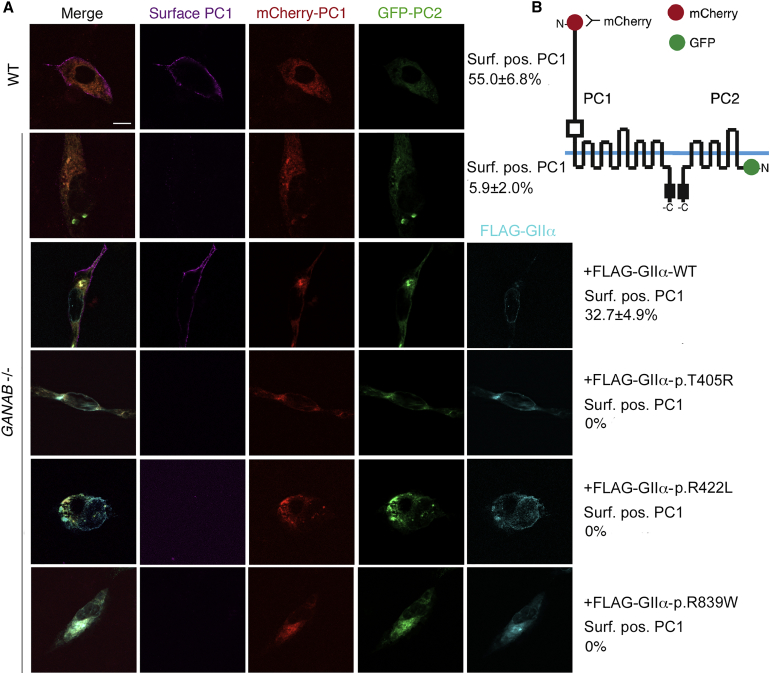
Autosomal-dominant polycystic kidney disease (ADPKD) is a common, progressive, adult-onset disease that is an important cause of end-stage renal disease (ESRD), which requires transplantation or dialysis. Mutations in PKD1 or PKD2 (∼85% and ∼15% of resolved cases, respectively) are the known causes of ADPKD. Extrarenal manifestations include an increased level of intracranial aneurysms and polycystic liver disease (PLD), which can be severe and associated with significant morbidity. Autosomal-dominant PLD (ADPLD) with no or very few renal cysts is a separate disorder caused by PRKCSH, SEC63, or LRP5 mutations. After screening, 7%-10% of ADPKD-affected and ∼50% of ADPLD-affected families were genetically unresolved (GUR), suggesting further genetic heterogeneity of both disorders. Whole-exome sequencing of six GUR ADPKD-affected families identified one with a missense mutation in GANAB, encoding glucosidase II subunit α (GIIα). Because PRKCSH encodes GIIβ, GANAB is a strong ADPKD and ADPLD candidate gene. Sanger screening of 321 additional GUR families identified eight further likely mutations (six truncating), and a total of 20 affected individuals were identified in seven ADPKD- and two ADPLD-affected families. The phenotype was mild PKD and variable, including severe, PLD. Analysis of GANAB-null cells showed an absolute requirement of GIIα for maturation and surface and ciliary localization of the ADPKD proteins (PC1 and PC2), and reduced mature PC1 was seen in GANAB(+/-) cells. PC1 surface localization in GANAB(-/-) cells was rescued by wild-type, but not mutant, GIIα. Overall, we show that GANAB mutations cause ADPKD and ADPLD and that the cystogenesis is most likely driven by defects in PC1 maturation.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Torres V.E., Harris P.C., Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. - PubMed
-
- The European Polycystic Kidney Disease Consortium The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77:881–894. - PubMed
-
- Mochizuki T., Wu G., Hayashi T., Xenophontos S.L., Veldhuisen B., Saris J.J., Reynolds D.M., Cai Y., Gabow P.A., Pierides A. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. - PubMed
-
- Heyer C.M., Sundsbak J.L., Abebe K.Z., Chapman A.B., Torres V.E., Grantham J.J., Bae K.T., Schrier R.W., Perrone R.D., Braun W.E., HALT PKD and CRISP Investigators Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2016 Published online January 28, 2016. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- U01 DK062401/DK/NIDDK NIH HHS/United States
- U01 DK056956/DK/NIDDK NIH HHS/United States
- P30 DK090728/DK/NIDDK NIH HHS/United States
- P30 DK090868/DK/NIDDK NIH HHS/United States
- U01 DK062402/DK/NIDDK NIH HHS/United States
- U01 DK056957/DK/NIDDK NIH HHS/United States
- U01 DK056943/DK/NIDDK NIH HHS/United States
- T32 DK007013/DK/NIDDK NIH HHS/United States
- U01 DK062408/DK/NIDDK NIH HHS/United States
- U01 DK056961/DK/NIDDK NIH HHS/United States
- U01 DK062411/DK/NIDDK NIH HHS/United States
- R01 DK044863/DK/NIDDK NIH HHS/United States
- U01 DK082230/DK/NIDDK NIH HHS/United States
- U01 DK062410/DK/NIDDK NIH HHS/United States
- R01 DK058816/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
