

Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits
- PMID: 27259154
- PMCID: PMC4893171
- DOI: 10.1016/j.cell.2016.04.048
Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits
Abstract
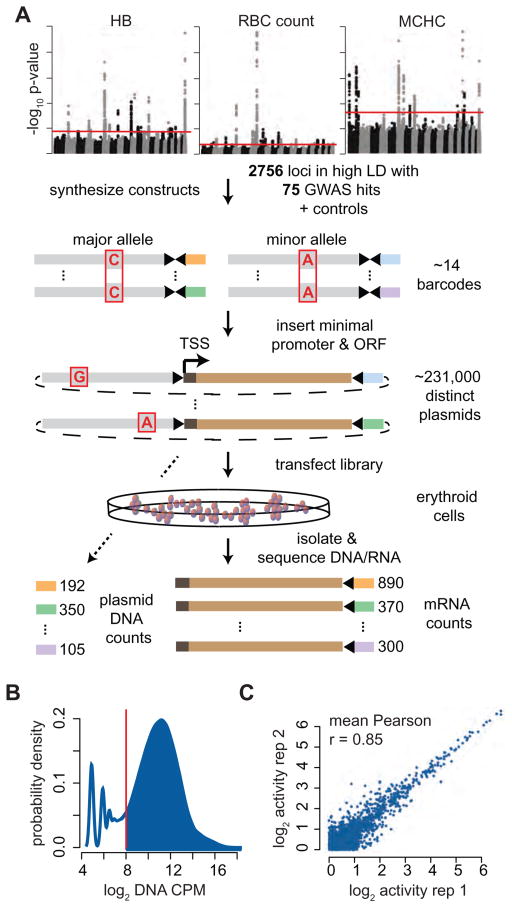
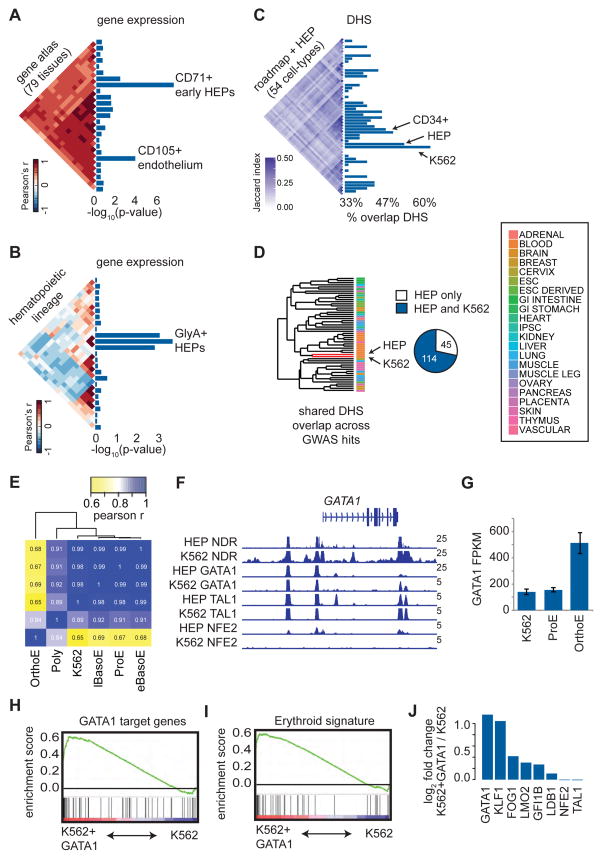
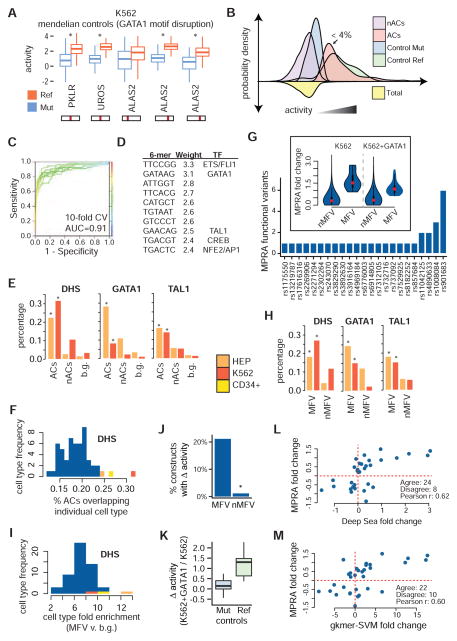
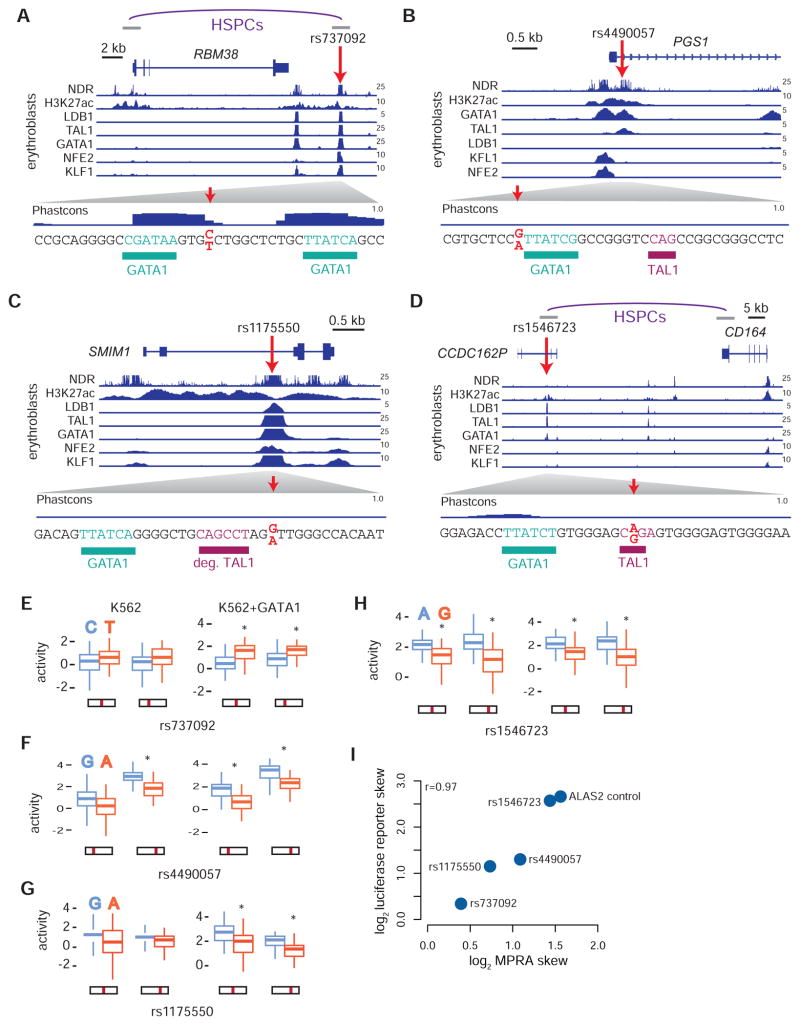
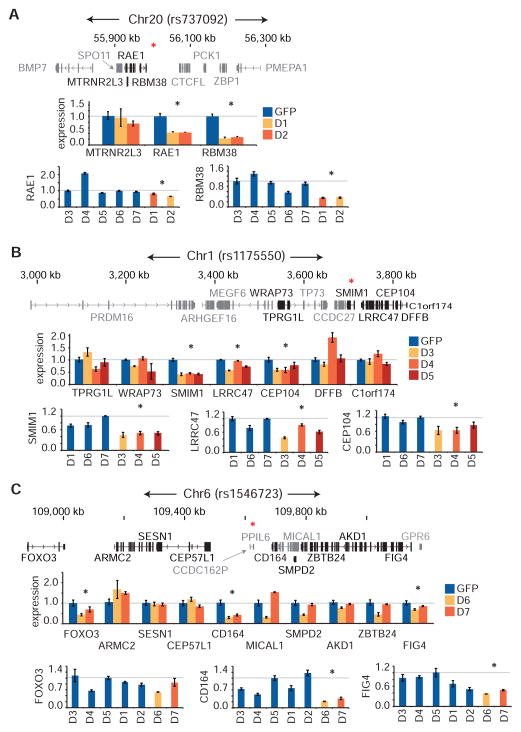
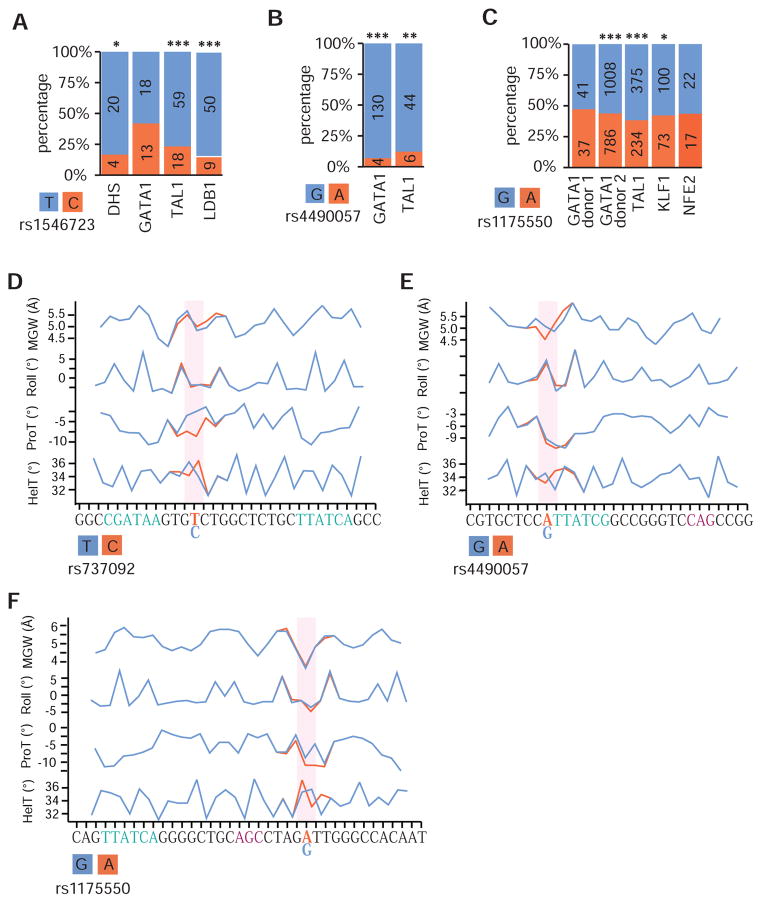
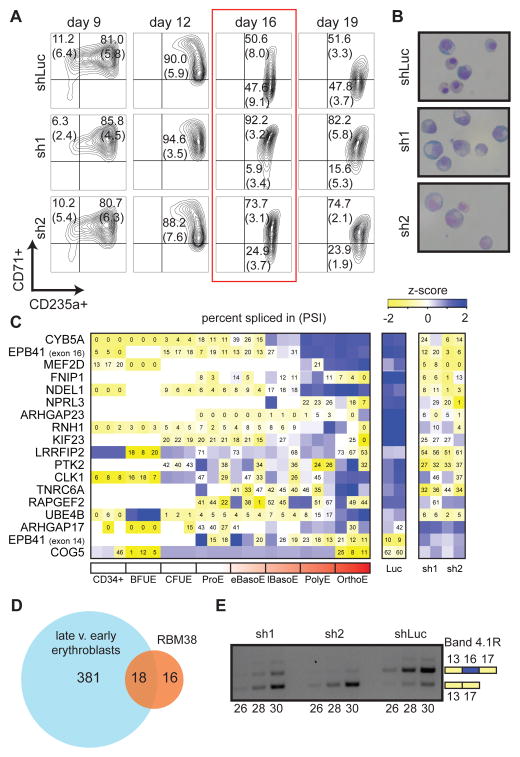
Genome-wide association studies (GWAS) have successfully identified thousands of associations between common genetic variants and human disease phenotypes, but the majority of these variants are non-coding, often requiring genetic fine-mapping, epigenomic profiling, and individual reporter assays to delineate potential causal variants. We employ a massively parallel reporter assay (MPRA) to simultaneously screen 2,756 variants in strong linkage disequilibrium with 75 sentinel variants associated with red blood cell traits. We show that this assay identifies elements with endogenous erythroid regulatory activity. Across 23 sentinel variants, we conservatively identified 32 MPRA functional variants (MFVs). We used targeted genome editing to demonstrate endogenous enhancer activity across 3 MFVs that predominantly affect the transcription of SMIM1, RBM38, and CD164. Functional follow-up of RBM38 delineates a key role for this gene in the alternative splicing program occurring during terminal erythropoiesis. Finally, we provide evidence for how common GWAS-nominated variants can disrupt cell-type-specific transcriptional regulatory pathways.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Campagna DR, de Bie CI, Schmitz-Abe K, Sweeney M, Sendamarai AK, Schmidt PJ, Heeney MM, Yntema HG, Kannengiesser C, Grandchamp B, et al. X-linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA-binding site mutations (vol 89, pg 315, 2014) Am J Hematol. 2014;89:670–670. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
