

Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome
- PMID: 27259756
- PMCID: PMC4958899
- DOI: 10.1093/brain/aww130
Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome
Abstract
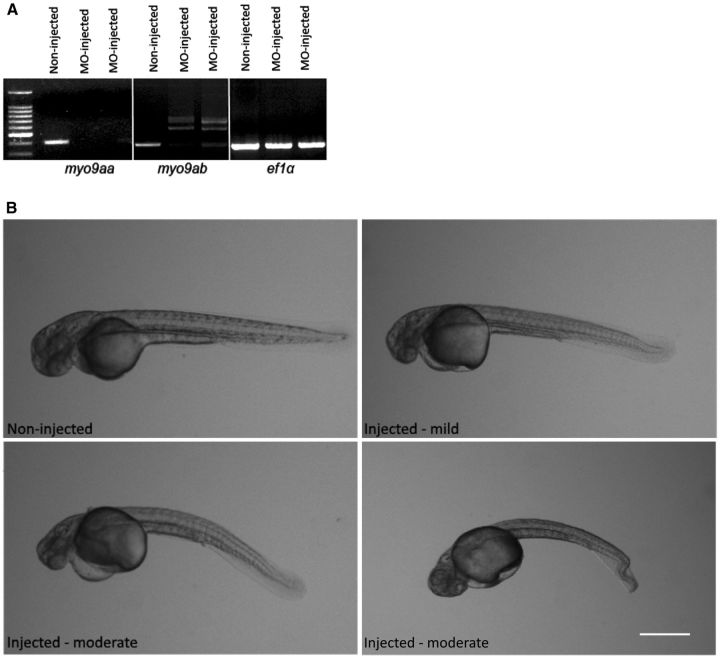
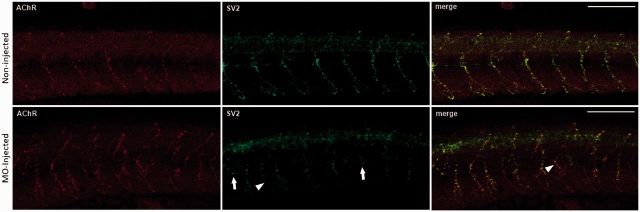
Congenital myasthenic syndromes are a group of rare and genetically heterogenous disorders resulting from defects in the structure and function of the neuromuscular junction. Patients with congenital myasthenic syndrome exhibit fatigable muscle weakness with a variety of accompanying phenotypes depending on the protein affected. A cohort of patients with a clinical diagnosis of congenital myasthenic syndrome that lacked a genetic diagnosis underwent whole exome sequencing in order to identify genetic causation. Missense biallelic mutations in the MYO9A gene, encoding an unconventional myosin, were identified in two unrelated families. Depletion of MYO9A in NSC-34 cells revealed a direct effect of MYO9A on neuronal branching and axon guidance. Morpholino-mediated knockdown of the two MYO9A orthologues in zebrafish, myo9aa/ab, demonstrated a requirement for MYO9A in the formation of the neuromuscular junction during development. The morphants displayed shortened and abnormally branched motor axons, lack of movement within the chorion and abnormal swimming in response to tactile stimulation. We therefore conclude that MYO9A deficiency may affect the presynaptic motor axon, manifesting in congenital myasthenic syndrome. These results highlight the involvement of unconventional myosins in motor axon functionality, as well as the need to look outside traditional neuromuscular junction-specific proteins for further congenital myasthenic syndrome candidate genes.
Keywords: MYO9A; congenital myasthenic syndrome; neuromuscular junction; unconventional myosin; whole exome sequencing.
© The Author (2016). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
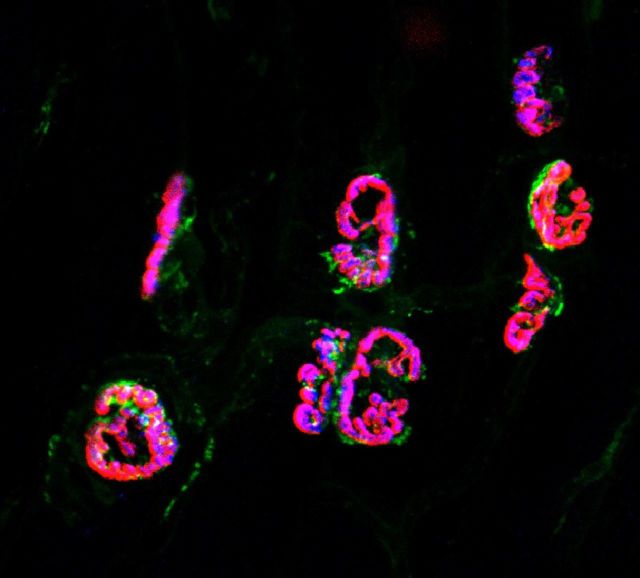
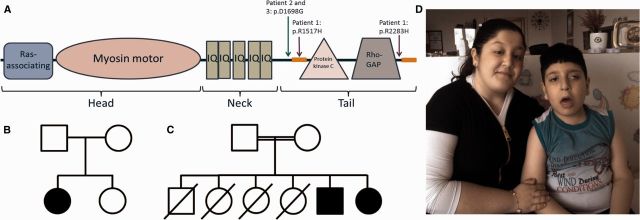
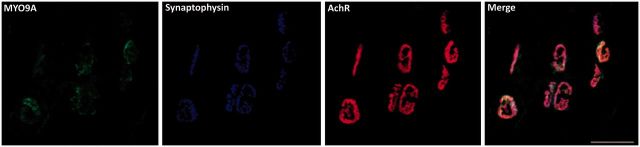
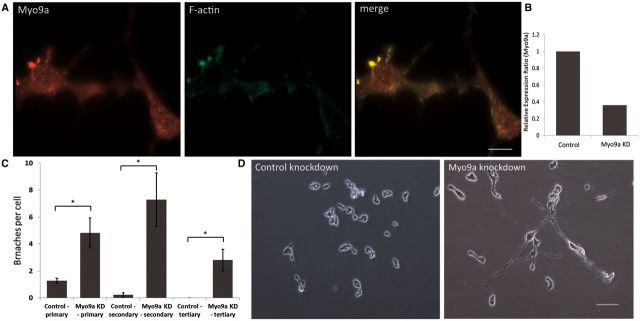
References
-
- Babin PJ, Goizet C, Raldua D. Zebrafish models of human motor neuron diseases: advantages and limitations. Prog Neurobiol 2014; 118: 36–58. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
