

GenFamClust: an accurate, synteny-aware and reliable homology inference algorithm
- PMID: 27260514
- PMCID: PMC4893229
- DOI: 10.1186/s12862-016-0684-2
GenFamClust: an accurate, synteny-aware and reliable homology inference algorithm
Abstract
Background: Homology inference is pivotal to evolutionary biology and is primarily based on significant sequence similarity, which, in general, is a good indicator of homology. Algorithms have also been designed to utilize conservation in gene order as an indication of homologous regions. We have developed GenFamClust, a method based on quantification of both gene order conservation and sequence similarity.
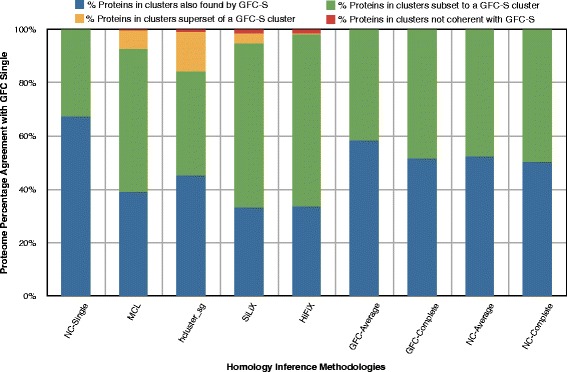
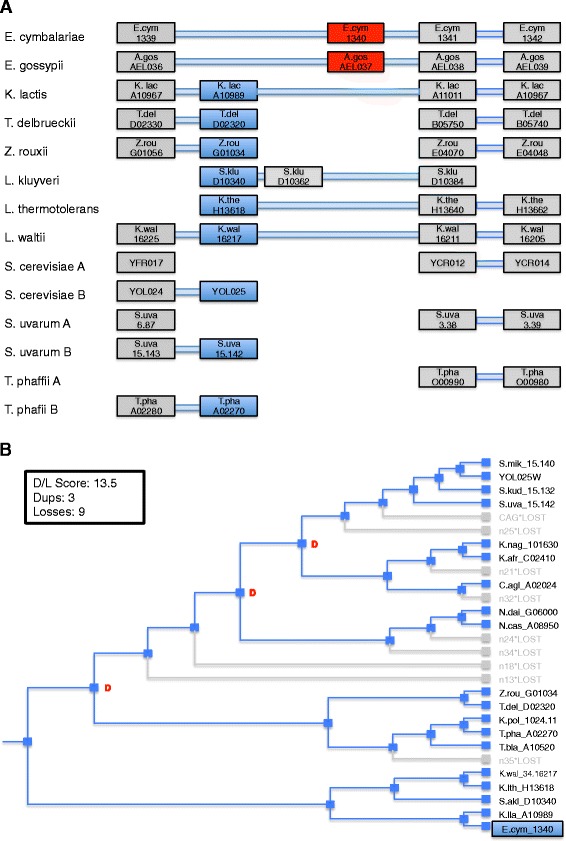
Results: In this study, we validate GenFamClust by comparing it to well known homology inference algorithms on a synthetic dataset. We applied several popular clustering algorithms on homologs inferred by GenFamClust and other algorithms on a metazoan dataset and studied the outcomes. Accuracy, similarity, dependence, and other characteristics were investigated for gene families yielded by the clustering algorithms. GenFamClust was also applied to genes from a set of complete fungal genomes and gene families were inferred using clustering. The resulting gene families were compared with a manually curated gold standard of pillars from the Yeast Gene Order Browser. We found that the gene-order component of GenFamClust is simple, yet biologically realistic, and captures local synteny information for homologs.
Conclusions: The study shows that GenFamClust is a more accurate, informed, and comprehensive pipeline to infer homologs and gene families than other commonly used homology and gene-family inference methods.
Keywords: Clustering; Gene family; Gene order conservation; Gene similarity; Gene synteny; Homology inference.
Figures









References
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
