

SMARCE1, a rare cause of Coffin-Siris Syndrome: Clinical description of three additional cases
- PMID: 27264197
- PMCID: PMC5870868
- DOI: 10.1002/ajmg.a.37722
SMARCE1, a rare cause of Coffin-Siris Syndrome: Clinical description of three additional cases
Abstract
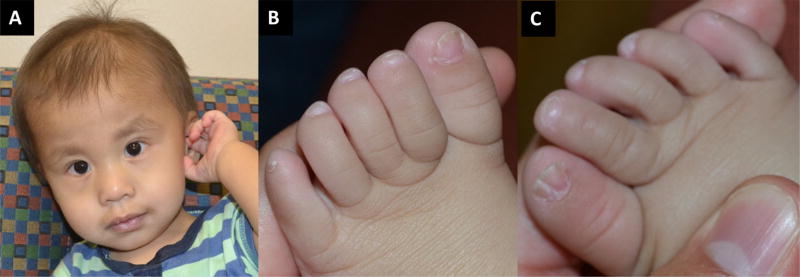
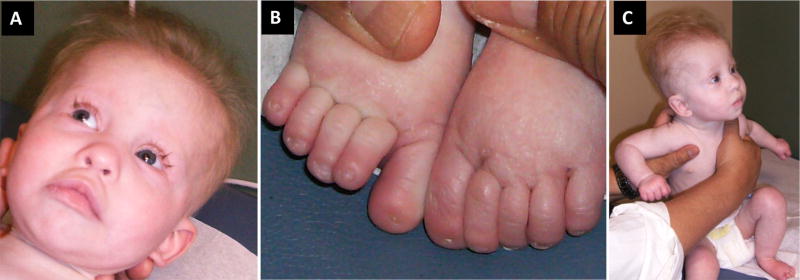
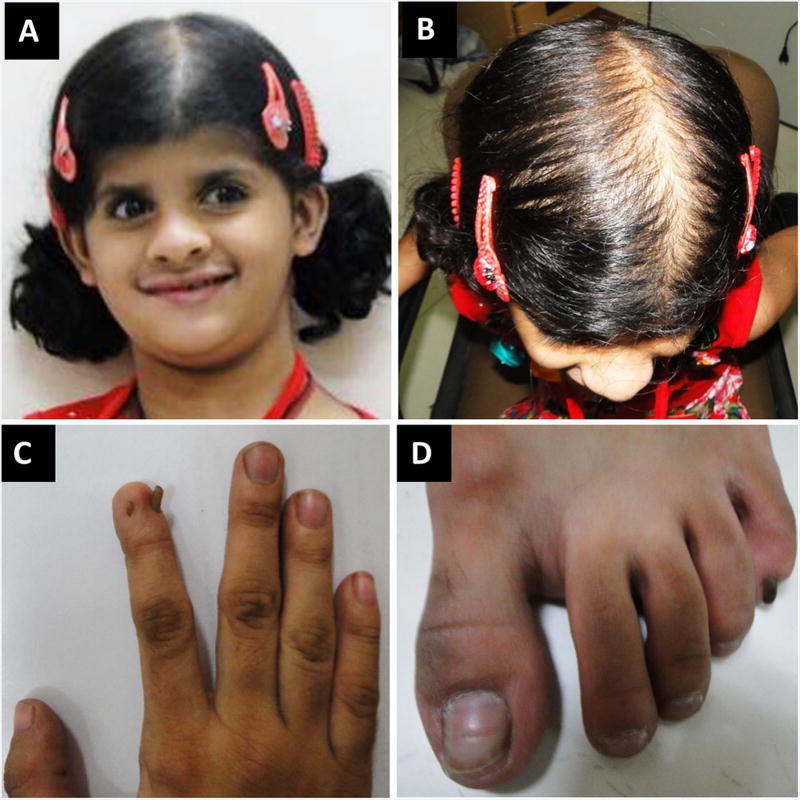
Coffin-Siris syndrome (CSS, MIM 135900), is a well-described, multiple congenital anomaly syndrome characterized by coarse facial features, hypertrichosis, sparse scalp hair, and hypo/aplastic digital nails and phalanges, typically of the 5th digits. Mutations in the BAF (SWI/SNF)-complex subunits (SMARCA4, SMARCE1, SMARCB1, SMARCA2, ARID1B, and ARID1A) have been shown to cause not only CSS, but also related disorders including Nicolaides-Baraitser (MIM 601358) syndrome and ARID1B-intellectual disability syndrome (MIM 614562). At least 200 individuals with CSS have been found to have a mutation in the BAF pathway. However, to date, only three individuals with CSS have been reported to have pathogenic variants in SMARCE1. We report here three additional individuals with clinical features consistent with CSS and alterations in SMARCE1, one of which is novel. The probands all exhibited dysmorphic facial features, moderate developmental and cognitive delay, poor growth, and hypoplastic digital nails/phalanges, including digits not typically affected in the other genes associated with CSS. Two of the three probands had a variety of different organ system anomalies, including cardiac disease, genitourinary abnormalities, feeding difficulties, and vision abnormalities. The 3rd proband has not had further investigative studies. Although an increasing number of individuals are being diagnosed with disorders in the BAF pathway, SMARCE1 is the least common of these genes. This report doubles the number of probands with these mutations, and allows for better phenotypic information of this rare syndrome. © 2016 Wiley Periodicals, Inc.
Keywords: BAF-complex; Coffin-Siris syndrome; SMARCE1; exome sequencing.
© 2016 Wiley Periodicals, Inc.
Conflict of interest statement
Conflicts of interest: none.
Figures
Similar articles
-
Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients.Hum Mutat. 2013 Nov;34(11):1519-28. doi: 10.1002/humu.22394. Epub 2013 Aug 30. Hum Mutat. 2013. PMID: 23929686
-
Numerous BAF complex genes are mutated in Coffin-Siris syndrome.Am J Med Genet C Semin Med Genet. 2014 Sep;166C(3):257-61. doi: 10.1002/ajmg.c.31406. Epub 2014 Jul 31. Am J Med Genet C Semin Med Genet. 2014. PMID: 25081545
-
Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A.Am J Med Genet C Semin Med Genet. 2014 Sep;166C(3):262-75. doi: 10.1002/ajmg.c.31407. Epub 2014 Aug 28. Am J Med Genet C Semin Med Genet. 2014. PMID: 25168959
-
Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature.Am J Med Genet A. 2013 Jun;161A(6):1221-37. doi: 10.1002/ajmg.a.35933. Epub 2013 May 1. Am J Med Genet A. 2013. PMID: 23637025 Review.
-
Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing.Am J Med Genet C Semin Med Genet. 2014 Sep;166C(3):241-51. doi: 10.1002/ajmg.c.31415. Epub 2014 Aug 28. Am J Med Genet C Semin Med Genet. 2014. PMID: 25169878 Review.
Cited by
-
BRG1 programs PRC2-complex repression and controls oligodendrocyte differentiation and remyelination.J Cell Biol. 2024 Jul 1;223(7):e202310143. doi: 10.1083/jcb.202310143. Epub 2024 Apr 23. J Cell Biol. 2024. PMID: 38652118 Free PMC article.
-
Landscape of mSWI/SNF chromatin remodeling complex perturbations in neurodevelopmental disorders.Nat Genet. 2023 Aug;55(8):1400-1412. doi: 10.1038/s41588-023-01451-6. Epub 2023 Jul 27. Nat Genet. 2023. PMID: 37500730 Free PMC article.
-
Coffin-Siris Syndrome and SMARCB1 Mutation Presenting With Schwannomatosis: A Case Report and Literature Review.Cureus. 2024 Aug 20;16(8):e67333. doi: 10.7759/cureus.67333. eCollection 2024 Aug. Cureus. 2024. PMID: 39170644 Free PMC article.
-
Mutational Landscapes and Phenotypic Spectrum of SWI/SNF-Related Intellectual Disability Disorders.Front Mol Neurosci. 2018 Aug 3;11:252. doi: 10.3389/fnmol.2018.00252. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30123105 Free PMC article. Review.
-
BICRA, a SWI/SNF Complex Member, Is Associated with BAF-Disorder Related Phenotypes in Humans and Model Organisms.Am J Hum Genet. 2020 Dec 3;107(6):1096-1112. doi: 10.1016/j.ajhg.2020.11.003. Epub 2020 Nov 23. Am J Hum Genet. 2020. PMID: 33232675 Free PMC article.
References
-
- Bhoj EJ, Li D, Harr MH, Tian L, Wang T, Zhao Y, Qiu H, Kim C, Hoffman JD, Hakonarson H, Zackai EH. Expanding the SPECC1L mutation phenotypic spectrum to include Teebi hypertelorism syndrome. Am J Med Genet Part A. 2015;167A:2497–2502. - PubMed
-
- Coffin GS, Siris E. Mental retardation with absent fifth fingernail and terminal phalanx. Am J Dis Child. 1970;119:433–439. - PubMed
-
- Fleck BJ, Pandya A, Vanner L, Kerkering K, Bodurtha J. Coffin-Siris syndrome: Review and presentation of new cases from a questionnaire study. Am J Med Genet. 2001;99:1–7. - PubMed
-
- Hempel A, Pagnamenta AT, Blyth M, Mansour S, McConnell V, Kou I, Ikegawa S, Tsurusaki Y, Matsumoto N, Lo-Castro A, Plessis G, Albrecht B, Battaglia A, Taylor JC, Howard MF, Keays D, Sohal S, DDD collaboration. Kühl SJ, Kini U, McNeill A. Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin-Siris syndrome. J Med Genet. 2016;53:152–162. - PMC - PubMed
-
- Kosho T, Okamoto N Coffin-Siris Syndrome International C. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet Part C Semin Med Genet. 2014;166C:262–275. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
